
Xeroderma pigmentosa: a report of three paediatric cases in Eastern Zambia, and review of the literature of this rare condition amongst sub-Saharan Africans
Derby Tembo, Suzanna Mwanza, Mbinga Mbinga, Chanda Kapoma, Malan Malumani
Corresponding author: Malan Malumani, Livingstone Central Hospital, Livingstone City, Southern Province, Zambia 
Received: 11 Aug 2021 - Accepted: 27 Sep 2022 - Published: 29 Sep 2022
Domain: Dermatology,Family Medicine,Pediatric ophthalmology
Keywords: Xeroderma pigmentosa, Cockayne syndrome, photosensitivity, case report
©Derby Tembo et al. PAMJ Clinical Medicine (ISSN: 2707-2797). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Derby Tembo et al. Xeroderma pigmentosa: a report of three paediatric cases in Eastern Zambia, and review of the literature of this rare condition amongst sub-Saharan Africans. PAMJ Clinical Medicine. 2022;10:15. [doi: 10.11604/pamj-cm.2022.10.15.31167]
Available online at: https://www.clinical-medicine.panafrican-med-journal.com//content/article/10/15/full
Case report 

Xeroderma pigmentosa: a report of three paediatric cases in Eastern Zambia, and review of the literature of this rare condition amongst sub-Saharan Africans
Xeroderma pigmentosa: a report of three paediatric cases in Eastern Zambia, and review of the literature of this rare condition amongst sub-Saharan Africans
![]() Derby Tembo1,
Derby Tembo1, ![]() Suzanna Mwanza1,
Suzanna Mwanza1, ![]() Mbinga Mbinga1,2,
Mbinga Mbinga1,2, ![]() Chanda Kapoma3,4,
Chanda Kapoma3,4, ![]() Malan Malumani3,4,&
Malan Malumani3,4,&
&Corresponding author
The diagnosis and management of xeroderma pigmentosa (XP) remains a challenge in sub-Saharan Africa due to limited health-care access, lack of specialist care and weak diagnostic testing. The purpose of this paper is to highlight the importance of clinical approach in the diagnosis of XP in a low-resource setting and to discuss some of the challenges these patients face living with XP. Three cases of children who presented with signs and symptoms of XP are discussed; their case histories are notable for demonstrating some of the challenges of managing the condition in sub-Saharan Africa. Management of XP is particularly complicated by the fact that there is no known cure for the condition and the need for complete shielding from ultraviolet (UV) light in a region with warm weather and abundant sunshine. A previously unreported effect of XP on the patients and their families is stigma and social exclusion. Setting up and linking the clients to psychosocial support groups would be of great benefit in alleviating the psychological effects of the disease.

Xeroderma pigmentosa (XP) is a rare inherited autosomal recessive disorder characterized by photosensitivity, pigmentary changes, premature skin aging, premalignant and malignant skin cancer development. It results from the defect in deoxyribonucleic acid (DNA) repair from ultraviolet (UV) induced damage [1]. Xeroderma pigmentosa (XP) can be divided into 8 subtypes (group A to G and a variant type V), each with a known responsible gene.
There is wide variation in the regional prevalence of the XP. The prevalence of the disease is estimated to be relatively high in the Middle East and Africa, however data is limited [2]. Global epidemiologic data reports at least one per million people affected in the United States, 2.3 per million in Western Europe, and 45 per million in Japan [3].
Clinically, XP has been classified into 3 groups: XP cutaneous disease where a patient only has cutaneous manifestations; secondly XP neurological disease with both cutaneous and neurological manifestations and lastly xeroderma pigmentosa/Cockayne syndrome (XP/CS) complex in which a patient develops both cutaneous and Cockayne syndrome manifestations [4]. The initial presenting signs in an affected child can range from extreme sensitivity to sunlight, triggering severe sunburn or abnormal lentiginosis; or freckle-like pigmentation due to increased numbers of melanocytes on sun-exposed areas. If affected individuals are not protected from sunlight, these symptoms will progress to increased or decreased pigmentation, skin aging and multiple skin cancers at an early age. UV light also causes anterior ocular segment lesions which include conjunctival xerosis and corneal drying, conjunctivitis, keratitis, evagination, corneal ulcers and decreased lacrimation. Additionally, up to 30% of patients worldwide show progressive neurodegenerative complications that greatly reduces the quality of life not only for the patient but also their families [4-6].
The diagnostic tests for XP include a photosensitivity test, DNA repair test using cultured fibroblasts and genetic analysis using peripheral blood or patient-derived cultured cells. These investigations are costly and not readily available in sub-Saharan Africa which makes clinical diagnosis the mainstay for XP. Hence the importance of standardizing clinical algorithms for diagnosing this condition in low- and middle-income settings. We report three cases of children who presented with signs and symptoms consistent with XP at Chipata Central Hospital (CCH), a third-level referral hospital for Eastern Zambia. The case histories are notable for demonstrating some of the challenges of managing the condition in sub-Saharan Africa. These include delayed diagnosis leading to advanced stages of the disease at diagnosis, difficulties in acquiring appropriate UV protective materials, challenges of complete sun protection in a region with warm weather and abundant sunlight, and resultant poor prognosis of the disease.
The diagnosis of these cases was made based on symptoms of chronic and acute photosensitivity, presence of skin cancer on sun-exposed areas and progressive neurodegenerative symptoms of unknown origin at the time of presentation as described by Moriwaki et al. [4].

Case 1
Patient information: SL a 4-year-old male presented to CCH with white spots in both eyes for approximately 2 years and generalised whitish skin blemishes from the age of 3 months. The parents reported that he was well until the age of 3 months when he developed hypochromic macula lesions on both arms, described as itchy when exposed to the sun. The lesions did not resolve with topical antifungal creams, prescribed at the local health facility. Later similar lesions appeared on the lower limbs, scalp and face extending to upper third of the trunk. Around the same time, dark eruption appeared on the face and scalp progressively increasing in size with some falling off spontaneously. At the age of 8 months, he started experiencing itchiness in both eyes with reddening and squinting associated with aversion to sunlight. Later the mother noted some whitish spots in both eyes, which did not improve with antibiotic eye drops. The past medical and developmental history were unremarkable. Family history available was limited, however, the child´s paternal half-uncle was reported to have had similar skin lesions and is presented in case 2.
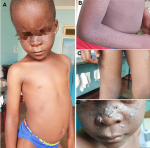
Clinical findings: on examination, there was freckling poikiloderma and xerosis of the skin involving the face, hands, upper third part of the trunk and lower limbs sparing the feet. There was also scaling, crusting and some nodular lesions on the nose as seen in Figure 1. Ophthalmological examination revealed photophobia, decreased visual acuity left eye and a conjunctival growth, described as nonspecific and crossing the limbus in the left eye about 2 cm to 3 cm on the corneal with neovascularisation. Examinations of the left eye fundus was not conclusive.
Diagnostic assessment: the diagnosis was based on the clinical findings described above. A skin biopsy confirmed the presence of a polypoid portion of skin-lined tissue with overlaying hyper and parakeratosis and extensive papillomatosis, without obvious dysplasia. Photosensitivity and genetic tests were not done due to limited resources.
Case 2
Patient information: SB a 6-year-old male presented to CCH with sores on the scalp for 3 years and progressively increasing papular lesions the face, tongue and nostrils. The patient was well until the age of one year, he began complaining of photophobia, pain and reddening of both eyes worsening with exposure to the sun. There was also a history of epistaxis associated with new lesions forming in both nostrils prior to presentation. The past medical and family were unremarkable.
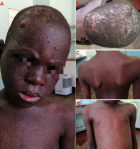
Clinical findings: clinically, he had multiple hyperkeratotic-papular lesions on the scalp that were approximately 1.0 cm x 2.0 cm in size. Ulcerative lesions were found on the lower lip and tongue with an erythematous base, ranging in size from 0.5 cm to 1 cm. He has hyper and hypo-pigmented macula lesions on the skin suggestive of poikiloderma with freckling as seen in Figure 2. Ophthalmological examination revealed visual acuity in the right eye at 6l/60 and marked limitation of the left eye, in which only large range movements were detectable. On slit lamp examination, there were corneal scars with hyperaemic conjunctival in both eyes.
Diagnostic assessment: diagnosis was based on clinical findings described above. A histopathological examination of a skin biopsy of the ulcer on the lip showed features of an invasive well-differentiated squamous cell carcinoma (SCC). Photosensitivity and genetic testing was not done due to limited resources.
Case 3
Patient information: MT a 4-year-old female who presented to CCH with malaria for which she was appropriately treated. The care givers reported hypopigmented skin on the limbs and ulcers on the lips, tongue and scalp from the age of 3 months. The lesions worsened with sun exposure. There was a history of cognitive and behaviour impairment where the child was only able to count up to 3 and had difficulties following instructions suitable for the age. Her behaviour was described as not being appropriate for age. There was a notable history of consanguinity.
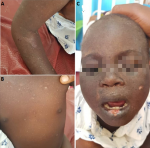
Clinical findings: on physical examination, the patient was small for age with weight and height less than 3rd percentile. She had hypopigmented macula lesions with freckling as seen in Figure 3. There were healing ulcerative lesions on the lower lip, anterior third of the tongue and on the scalp. Ophthalmological examination revealed bilateral corneal opacities with conjunctival hyperaemia and photosensitivity.
Diagnostic assessment: it was based on the clinical findings; no confirmatory tests were done due to limited resources.
Therapeutic intervention: the 3 patients were prescribed a topical cream with a sun protection factor of 50 (SPF-50), the parents were educated on the importance of protection from direct sunlight by wearing long-sleeved clothing and use of sunglasses. SPF-50 was not always readily available in the hospital pharmacy and acquiring appropriate sunscreen clothing was another challenge.
Follow-up: the patients were scheduled for 3 monthly reviews, to be seen in consultation with the dermatologist to ensure adherence to the preventive measures and early detection of complications. It has been a challenge to comply with this because of long distance to the hospital and COVID-19 restriction measures.
Ethical consideration: informed verbal consent was obtained from the guardians of the patients and no identifying information was used in the document.
These cases represent several challenges in the care for children with XP in a rural region of sub-Saharan Africa. These includes limited healthcare access, lack of appropriate specialist care services, and weak diagnostic capacity. Additionally, once diagnosed, management of this incurable condition is complicated in our region. Not only is the climate one of intense long hours of sunlight, but there is limited availability of broad spectrum sunscreen protection with recommendable factor (SPF). Additionally, the average income in our region is low and is often dependent upon small-scale agriculture activities, and consequently unable to access adequate protective clothing for standard lifelong ultraviolet light protection, further complicating recommendations for minimizing sun exposure.
Unfortunately, the physical manifestations of XP also carry strong stigma in Zambia, which limits inclusion for children within community activities and school as is the case with children living with albinism [7]. The patient in case 1 is reported to have stopped attending pre-school because the friends at school were scared of his physical appearance, this negative behaviour towards the patient is reported to have reduced after the class teacher was advised to discuss the patients´ condition with the students. The patient in case 2 is not yet in school as he was advised to enrol at a school with special needs. Other authors have attributed varying degrees of neurological deficits such as cognitive deficits to the high dropout rate from schools as it is the case with our patients [5,8,9]. The eye manifestations in these patients also poses a huge challenge in these patients during their daily community and school activities.
Management of these patients require a holistic multidisciplinary approach which should be inclusive of health care workers from community health care workers to specialist clinicians which include scheduled reviews, visiting their homes to ensure adherence to basic UV protection and creating awareness in the community and schools these children attend. These patients need to be given social, financial and psychological support which will help to improve adherence to standard UV protective measures.
The prevalence of XP in Zambia and the sub-Saharan Africa is unknown, sporadic cases have been reported in many countries for which there is need for an epidemiological study to determine the prevalence of XP in the region. This will help with policy planning for the development of structural systems in order to improve care and survival of these children.
Zambia´s healthcare system exemplifies a developing system in which there are wide disparities in the health care capacities between urban and rural areas. In our case, the patients were co-managed in consultation with a dermatologist based in another region of the country via mobile based internet applications (i.e. WhatsApp messaging and phone calls). Developing strong e-health services such as telemedicine, can play a significant role in improving access to specialist care by rural communities thereby improving care outcomes among the paediatric populations. Telemedicine and telehealth are becoming an important integral aspect in the care of patients especially in human resource deficient and rural health care setups like in Zambia [10,11].
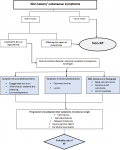
Currently, there is no cure for XP, care for these patients include measures that ensures complete shielding from UV and symptomatic treatment for complications. The measures against UV include application of a sun screen formulation with a high sun protection factor (SPF), wearing of protective clothing and use of special glasses for any outdoor activity or when going for school. There is also need to provide coverings on windows such as film coating or curtains, of homes and classrooms of children with XP in order to limit the exposure to UV light even when in enclosed spaces. Measures against skin cancer include frequent reviews with a dermatologist to check if the sun protection is effective and examination for early detection of skin cancers. These patients also need an ophthalmological checkup every 3-6 months for eye manifestations of XP [4]. Early stimulation at home and school is encouraged to promote neurological development. We propose a clinical diagnostic screening tool for diagnosis of XP in rural areas in order to aid quick recognition, appropriate and timely referral of these patients (Figure 4).
Early diagnosis and management of children with XP, in a multidisciplinary approach could improve survival of these patients even in resource-limited settings. In the absence of appropriate laboratory confirmation of the condition a clinical diagnostic algorithm can be valuable in aiding timely recognition of the disease. Strong e-health systems are absolute necessities in improving care outcomes even for rare conditions such as XP. Psychosocial care of the children and their families is necessary for social inclusion of these patient.
The authors declare no competing interests.
Derby Tembo, Suzanna Mwanza, Mbinga Mbinga, Chanda Kapoma, and Malan Malumani were in direct contact and management of patients; Derby Tembo and Malan Malumani did the initial draft of article and Suzanna Mwanza, Mbinga Mbinga and Chanda Kapoma did the revision and edits; all authors were involved in the writing and case management of the patients. All the authors read and approved the final version of this manuscript.
Figure 1: A,B,C) freckling poikiloderma and xerosis of the skin involving the face, hands, upper third part of the trunk and lower limbs sparing the feet; D) scaling, crusting and some extensive papillomatosis nodular lesions on the nose
Figure 2: A) multiple hyperkeratotic-papular lesions on the scalp that were approximately 1.0 cm x 2.0 cm; B) ulcerative lesions were found on the lower lip and tongue with an erythematous base, ranging in size from 0.5cm to 1cm; C,D) hyper and hypo-pigmented macula lesions on the skin suggestive of poikiloderma with freckling
Figure 3: A,B) hypopigmented macula lesions with freckling; C) healing ulcerative lesions on the lower lip, anterior third of the tongue
Figure 4: proposed clinical diagnostic screening tool for diagnosis of XP in low resource centres in order to aid quick recognition, appropriate and timely referral of XP patients
- Butt FM, Moshi JR, Owibingire S, Chindia ML. Xeroderma pigmentosum: a review and case series. J Craniomaxillofac Surg. 2010 Oct;38(7):534-7. PubMed | Google Scholar
- Black JO. Xeroderma pigmentosum. Head Neck Pathol. 2016 Jun;10(2):139-44. PubMed | Google Scholar
- Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum: cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol. 1987 Feb;123(2):241-50. PubMed | Google Scholar
- Moriwaki S, Kanda F, Hayashi M, Yamashita D, Sakai Y, Nishigori C. Xeroderma pigmentosum clinical practice guidelines. J Dermatol. 2017 Oct;44(10):1087-1096. PubMed | Google Scholar
- Lehmann AR, McGibbon D, Stefanini M. Xeroderma pigmentosum. Orphanet J Rare Dis. 2011 Nov 1;6:70. PubMed | Google Scholar
- Kaloga M, Dioussé P, Diatta BA, Bammo M, Kourouma S, Diabate A et al. Squamous cell carcinoma in African children with xeroderma pigmentosum: three case reports. Case Rep Dermatol. 2016 Nov 15;8(3):311-318. PubMed | Google Scholar
- Rohwerder B. Disability stigma in developing countries. 2018. Google Scholar
- Natale V, Raquer H. Xeroderma pigmentosum-Cockayne syndrome complex. Orphanet J Rare Dis. 2017 Apr 4;12(1):65. PubMed | Google Scholar
- Kraemer KH, DiGiovanna JJ, Tamura D. Xeroderma pigmentosum. In: Adam MP, Ardinger HH, Pagon RA, et al. (eds) GeneReviews�. Seattle (WA): University of Washington, Seattle, 1993-2022. PubMed
- Combi C, Pozzani G, Pozzi G. Telemedicine for developing countries. a survey and some design issues. Appl Clin Inform. 2016 Nov 2;7(4):1025-1050. PubMed | Google Scholar
- Nelson R. Telemedicine and telehealth: the potential to improve rural access to xare. Am J Nurs. 2017 Jun;117(6):17-18. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures
Article metrics

