Révélation atypique digestive de la granulomatose éosinophilique avec polyangéïte (syndrome de Churg et Strauss): à propos d´un cas
Mariam Konso, Hicham El Bacha, Fatimazahra Mghyly, Salma Douihi Touzani, Imane El Hamraoui, Zahira Hazil, Abdelkrim Boulanouar, Amina Berraho, Nadia Benzzoubeir, Errabih Ikram
Corresponding author: Mariam Konso, Service d'Hépato-Gastro-Entérologie et Proctologie « Médecine B », Hôpital Ibn Sina, CHU Ibn Sina, Université Mohamed V, Souissi, Rabat, Maroc 
Received: 26 Jul 2021 - Accepted: 02 Nov 2023 - Published: 07 Dec 2023
Domain: Gastroenterology
Keywords: Vascularite à ANCA, syndrome de Churg-Strauss, hyperéosinophilie, cas clinique
©Mariam Konso et al. PAMJ Clinical Medicine (ISSN: 2707-2797). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Mariam Konso et al. Révélation atypique digestive de la granulomatose éosinophilique avec polyangéïte (syndrome de Churg et Strauss): à propos d´un cas. PAMJ Clinical Medicine. 2023;13:31. [doi: 10.11604/pamj-cm.2023.13.31.30940]
Available online at: https://www.clinical-medicine.panafrican-med-journal.com//content/article/13/31/full
Case report 

Révélation atypique digestive de la granulomatose éosinophilique avec polyangéïte (syndrome de Churg et Strauss): à propos d´un cas
Révélation atypique digestive de la granulomatose éosinophilique avec polyangéïte (syndrome de Churg et Strauss) : à propos d'un cas
Atypical digestive manifestations of eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome): a case report
Mariam Konso1, ![]() Hicham El Bacha1, Fatimazahra Mghyly1,
Hicham El Bacha1, Fatimazahra Mghyly1, ![]() Salma Douihi Touzani1, Imane El Hamraoui1,
Salma Douihi Touzani1, Imane El Hamraoui1, ![]() Zahira Hazil2, Abdelkrim Boulanouar2, Amina Berraho2, Nadia Benzzoubeir1, Errabih Ikram1
Zahira Hazil2, Abdelkrim Boulanouar2, Amina Berraho2, Nadia Benzzoubeir1, Errabih Ikram1
&Auteur correspondant
La granulomatose Éosinophilique avec polyangéïte (EGPA) appelée syndrome de Churg-Strauss est une vascularite nécrosante touchant les vaisseaux de petit calibre. Elle se caractérise par de l'asthme et une hyperéosinophilie associés à des manifestations extrapulmonaires. Des manifestations digestives peuvent être observées, conditionnant le pronostic et peuvent mimer en tous points les maladies inflammatoires chronique de l'intestin. Les anticorps Anticytoplasme des polynucléaires neutrophiles (ANCA) sont positifs dans la moitié des cas type anti-myélopéroxidase (MPO). Nous rapportons le cas d'une patiente hospitalisée au Service de Gastro-Entérologie (Médecine B) au Centre Hospitalier Universitaire Ibn-Sina à Rabat pour EGPA révélée par une atteinte digestive et ophtalmologique. Nous rappellerons les principales données cliniques et paracliniques de ce syndrome dans la littérature.
Eosinophilic granulomatosis with polyangiitis (EGPA) or Churg-Strauss syndrome is a necrotizing vasculitis affecting small vessels. It is characterised by the association between asthma and hypereosinophilia with extrapulmonary manifestations. Digestive manifestations may be observed, which affect prognosis and may mimic chronic inflammatory bowel diseases. Patients are positive for Antineutrophil cytoplasmic antibodies (ANCA), directed against Myeloperoxidase (MPO) in half of the cases. We here report the case of a patient hospitalized with EGPA revealed by digestive and ophthalmological involvement in the Division of Gastroenterology (Medicine B) at the Ibn-Sina University Hospital Center in Rabat. The purpose of this study was to review the main clinical and paraclinical data on this syndrome in the literature.
Key words: ANCA-associated vasculitis, Churg-Strauss syndrome, hypereosinophilia, case report

La granulomatose Éosinophilique avec polyangéïte (EGPA) appelée syndrome de Churg et Strauss est une vascularite nécrosante touchant les vaisseaux de petit calibre. Sur le plan clinique elle se caractérise par un asthme et des signes systémiques extrapulmonaires notamment l'atteinte digestive et ophtalmologique qui engage le pronostic. Nous rappellerons les principales données cliniques et paracliniques de ce syndrome dans la littérature à travers un cas clinique suivi au Service de Gastro-Entérologie (Médecine B) au Centre Hospitalier Universitaire Ibn-Sina à Rabat.

Informations relatives au patient: il s'agit d'une patiente âgée de 56 ans ayant un asthme sous salbutamol inhalé depuis 30 ans, admise pour des douleurs abdominales et des diarrhées glaireuses.
Résultats cliniques: sur le plan clinique on note une sensibilité abdominale diffuse à la palpation.
Chronologie: la symptomatologie remonte à un mois de son admission par l'installation de douleurs abdominales diffuses, continues et aggravées par l'alimentation, associées à des vomissements alimentaires et des diarrhées glaireuses, le tout évoluant dans un contexte d'altération de l'état général et d'amaigrissement chiffré à 10 kg. Par ailleurs, la patiente rapporte également une notion d'arthralgies depuis 6 mois. L'évolution a été marquée par une baisse de l'acuité visuelle brutale à prédominance droite.
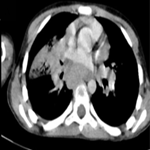
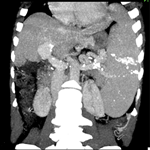
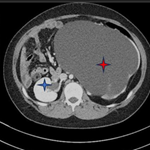
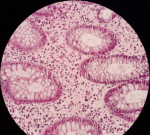
Démarche diagnostique: sur le plan radiologique, le scanner abdominal objective un épaississement inflammatoire diffus intéressant l'estomac, les anses iléales et le colon sigmoïde étendu jusqu'au mi-colon transverse pouvant être en rapport avec une maladie inflammatoire chronique de l'intestin (Figure 1). Biologiquement, l'hémogramme montre une anémie normochrome normocytaire d'origine inflammatoire avec une hémoglobine à 9g/dl, une hyperleucocytose à 18840mm³, une hyperéosinophilie à 10090/mm³ et des plaquettes à 272000/mm³. La proteine C réactive élevée à 98,20 mg/l. Le test QuantiFERON était négatif et le dosage sanguin des marqueurs tumoraux était normal. La coloscopie totale retrouve une sténose du colon transversée infranchissable par une muqueuse érythémateuse au niveau colique gauche siège de multiples érosions. Sur le plan histologique, l'examen anatomopathologique des biopsies coliques retrouve une muqueuse colique présentant une ulcération comblée par un tissu de granulation et abritant des cellules éosinophiles (Figure 2).
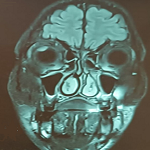
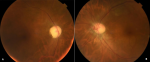
L'examen ophtalmologique objective une acuité visuelle réduite à perception lumineuse réduite en œil droit et 3/10 en œil gauche avec une motilité oculaire conservée et un réflexe photomoteur direct et consensuel altéré en œil droit, le tonus oculaire ainsi que l'examen du segment antérieur étaient sans particularités. Au fond d'œil, l'examen objective une papille pâle en œil droit et un aspect d'œdème sectoriel supéro-nasal en œil gauche. Une angio-imagerie par résonance magnétique cérébrale a été réalisée et est revenue sans anomalies. La tomographie en cohérence optique papillaire a confirmé l'atrophie papillaire. L'examen ophtalmologique est en faveur d'une neuropathie optique antérieure compliquée d'atrophie optique, et dont l'hypothèse ischémique est la plus probable (Figure 3). La recherche d'autoanticorps antineutrophiles cytoplasmiques était positive à 95 de type anti-MPO. Le diagnostic d'EGPA était donc retenu sur l'association d'un asthme avec hyperéosinophilie sanguine, de manifestations systémiques, digestive et ophtalmologique. Le score pronostique Five factor score (FFS) à 1.
Intervention thérapeutique: un traitement médical a été instauré, à base de corticothérapie initialement par voie intra veineuse (3 bolus de méthylprednisolone à 15mg/kg), puis per os (prednisone 1mg/kg/j).
Suivi et résultats des interventions thérapeutiques: l'amélioration clinique et biologique était rapide avec normalisation immédiate de l'éosinophilie et régression des douleurs abdominale après quelques jours de traitement, puis la patiente a été transférée dans un service de médecine interne pour le suivie de sa maladie.
Consentement éclairé: le patient a donné son consentement éclairé.
La granulomatose éosinophilique avec polyangéite (EGPA) est une vascularite systémique et pulmonaire qui fut individualisée de la périartérite noueuse en 1951 par Jacob Churg et Lotte Strauss dans une publication princeps de 13 observations [1]. La EGPA se manifeste généralement entre 7 et 74 ans, avec un âge moyen d'apparition de 38 à 54 ans [2]. Aucune prédominance de sexe ou de prédisposition ethnique n'a été clairement démontrée dans la EGPA [3]. Habituellement l'asthme précède les manifestations systémiques et les malades ont une sinusite maxillaire et souvent des antécédents allergiques. Churg et Strauss notèrent trois critères histologiques permettant de faire le diagnostic [1]: une infiltration tissulaire par des éosinophiles, une vascularite nécrosante et des granulomes extravasculaires. Les lésions histologiques caractéristiques de la EGPA sont la vascularite et le granulome extravasculaire. La vascularite peut être ou non granulomateuse et il ne faut pas s'attendre à rencontrer une forme histologique complète dans tous les cas de EGPA. Le granulome est inflammatoire avec présence des cellules géantes [4].
Plusieurs mécanismes contribuent au développement de la EGPA. Un terrain prédisposant est certainement présent comme la présence d'un asthme chez le patient ou sa famille, des antécédents d'allergie telle une rhinite ou des sinusites récidivantes. Sur ce terrain prédisposé, la EGPA survient de façon inopinée ou la suite de circonstances déclenchant la maladie comme des vaccinations ou des désensibilisations [5]. La maladie est associée dans un certain nombre de cas aux ANCA, essentiellement de type MPO. Ces ANCA sont pathogènes [6]. Ce mécanisme pourrait rendre compte d'un certain nombre de manifestations cliniques, en particulier celles en rapport avec la capillarite ou l'atteinte glomérulaire. Lanham et al. [7] ont décrit une histoire naturelle de EGPA évoluant en 3 phases. Une première phase dite « allergique » (asthme, rhinite allergique), une seconde dite « hyperéosinophilique » (augmentation des polynucléaires éosinophiles dans le sang) et enfin une phase de « vascularite » à proprement dit ou l'atteinte systémique s'installe rapidement, en quelques semaines ou tout au plus deux trois mois, dans un contexte de gravité clinique rapidement évident [8]. Les signes généraux pouvant être retrouvé sont une asthénie, une fièvre dans la moitié des cas et un amaigrissement. Il y a également fréquemment des douleurs articulaires mais pas d'arthrite, ni de déformation des articulations. Les manifestations pleuropulmonaires sont dominées par l'asthme au premier plan, débute relativement tardivement, vers l'âge de 30 ans et est souvent sévère et corticodépendante [4].
Les hémorragies alvéolaires, les pleurésies sont également décrites. Les neuropathies périphériques de topographie distale prédominant aux membres inférieurs sont les plus fréquentes des manifestations neurologiques de EGPA et très suggestives du diagnostic. L'atteinte la plus caractéristique est celle du nerf sciatique poplité externe et, un moindre degré, du nerf sciatique poplité interne [4]. L'atteinte digestive est un facteur de mauvais pronostic de EGPA [9]. Des formes de EGPA en apparence limitées au tube digestif ont été rapportées [10]. L'infiltration de la paroi intestinale par des polynucléaires éosinophiles aboutit parfois une entérocolite éosinophiles qui peut être très étendue. Les lésions siègent tout au long du tractus digestif estomac, intestin grêle, appendice, côlon, rectum qui peuvent mimer une maladie de Crohn, mais aussi dans la vésicule biliaire, sur le péritoine donnant une ascite riche en polynucléaires éosinophiles. La symptomatologie digestive est prédominée par les douleurs abdominales (30 à 60% des patients) et un moindre degré les nausées, les vomissements et la diarrhée. La survenue d'hémorragies et/ou de perforations intestinales, la persistance de douleurs abdominales intenses malgré le traitement et un amaigrissement sont des éléments cliniques de mauvais pronostic. Un méléna ou une hématémèse peuvent survenir [8].
Les explorations complémentaires n'apportent que peu de renseignements, individualisent des épaississements inflammatoires des parois des anses digestives, parfois évocateurs (mais non spécifique). Les endoscopies digestives ont surtout un intérêt pour le diagnostic différentiel, surtout que les vascularites à ANCA miment une atteinte digestive typique de la maladie de Crohn [8]. Les manifestations ophtalmologiques rapportées au cours de la EGPA sont des pseudotumeurs inflammatoires de l'orbite, des atteintes conjonctivales et neuro-ophtalmologiques, une neuropathie optique ischémique antérieure associée à une ischémie choroïdienne, une paralysie oculomotrice et des infarctus rétiniens. D'autres manifestations peuvent être observées, telles que l'atteinte cutanée (purpura vasculaire, livedo reticularis, syndrome de Raynaud), l'atteinte cardiaque qui engage le pronostic vital (péricardite, atteinte endomyocardique, arythmie), et également l'atteinte rénale qui est rare à type de glomérulonéphrites. L'hyperéosinophilie sanguine, l'élévation des IgE sériques et la présence d'ANCA anti-MPO constituent les trois principales anomalies biologiques. Un syndrome inflammatoire est habituel. L'hyperéosinophilie sanguine est un des arguments diagnostique de EGPA [5]. Sur la base des travaux de Churg et Strauss [1] plusieurs définitions et critères de classification pour le diagnostic du syndrome de Churg et Strauss [1]. Au fur et à mesure ces critères diagnostique ont évolué, et s'appliquent des patients chez qui le diagnostic de vascularite a été fait histologiquement [8].
Le pronostic dépend de l'extension anatomique de la maladie et de la nature des organes atteints. Le Five Factor Score (FFS) [9] peut être utilisé pour déterminer la stratégie thérapeutique (Tableau 1). Il est probable que les patients ayant un FFS égal 0, ne nécessitent qu'un traitement par corticoïdes. Quand le FFS est supérieur ou égal à 1, un traitement immunosuppresseur (essentiellement le cyclophosphamide Endoxan®) est indiqué en première intention. Le traitement est à base de corticoïdes (prednisone), la posologie est de l'ordre d'1mg/kg par jour peut être précédée par un trois bolus IV de méthylprednisolone consécutifs à la dose de 15 mg/kg par jour. Le traitement immunosuppresseur de choix est le cyclophosphamide (Endoxan®). La durée du traitement par cyclophosphamide est habituellement d'un an. Il est à noter que ce qu'il faut tirer de ce cas clinique, est de ne pas conclure rapidement à une maladie inflammatoire chronique de l'intestin et de garder en tête une atteinte digestive des vascularites.
La granulomatose éosinophile avec polyangéïte est une maladie rare, c'est une vascularite systémique sévère pouvant affecter tous les organes. Elle peut mimer en tout point une maladie de Crohn, l'atteinte digestive peut être sévère et de mauvais pronostic. Le traitement est à base de corticoïdes et d'immunosuppresseurs et doit être adapté à chaque patient en fonction de la présence de facteurs pronostiques.
Les auteurs ne déclarent aucun conflit d'intérêts.
Tous les auteurs ont contribué à la conduite de ce travail. Tous les auteurs déclarent également avoir lu et approuvé la version finale du manuscrit.
Figure 1: scanner avec injection de produit de contraste montrant un épaississement de la paroi du colon sigmoïde
Figure 2: aspect anatomopathologique de la biopsie colique
Figure 3: A) œil droit: rétine objectivant une pâleur papillaire; B) œil gauche: aspect d'œdème sectoriel
- Churg J, Strauss L. Allergic granulomatosis, allergic angiitis, and periarteritis nodosa. Am J Pathol. 1951 Mar-Apr;27(2):277-301. PubMed | Google Scholar
- Kahn JE, Bl�try O, Guillevin L. Hypereosinophilic syndromes. Best Pract Res Clin Rheumatol. 2008 Oct;22(5):863-82. PubMed
- Piram M, Maldini C, Mahr A. Effect of race/ethnicity on risk, presentation and course of connective tissue diseases and primary systemic vasculitides. Curr Opin Rheumatol. 2012 Mar;24(2):193-200. PubMed | Google Scholar
- Guillevin L, Pagnoux C, Cohen P. Syndrome de Churg-Strauss. Revue française d'allergologie et d'immunologie clinique. 2004;44:96-10. Google Scholar
- Guillevin L, Amouroux J, Arbeille B, Boura R. Churg-Strauss angiitis. Arguments favoring the responsibility of inhaled antigens. Chest. 1991 Nov;100(5):1472-3. PubMed | Google Scholar
- Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest. 2002 Oct;110(7):955-63. PubMed | Google Scholar
- Lanham JG, Elkon KB, Pusey CD, Hughes GR. Systemic vasculitis with asthma and eosinophilia: a clinical approach to the Churg-Strauss syndrome. Medicine (Baltimore). 1984 Mar;63(2):65-81. PubMed | Google Scholar
- Lhote F. Syndrome de Churg et Strauss. Presse Med. 2007 May;36(5 Pt 2):875-89. PubMed | Google Scholar
- Guillevin L, Lhote F, Gayraud M, Cohen P, Jarrousse B, Lortholary O, et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine (Baltimore). 1996 Jan;75(1):17-28. PubMed | Google Scholar
- Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Casassus P. Churg-Strauss syndrome. Clinical study and long-term follow-up of 96 patients. Medicine (Baltimore). 1999 Jan;78(1):26-37. PubMed | Google Scholar
Search

This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures
Article metrics
Recently from the PAMJ-CM