
Pediatric chronic myeloid leukemia: a case report of a disease with a unique biology
Abir Yahyaoui, Imane Douichi, Nada Ouahabi, Wissame Azizi, Abdessamad Amrani, Abdelilah Berhili, Nabiha Trougouty, Mounia Slaoui, Mohammed Bensalah, Ayad Ghanam, Rachid Seddik
Corresponding author: Abir Yahyaoui, Hematology Laboratory, Mohammed VI University Hospital, Faculty of Medicine and Pharmacy of Oujda, Mohammed First University, Oujda, Morocco 
Received: 15 Feb 2024 - Accepted: 05 Apr 2024 - Published: 16 Apr 2024
Domain: Laboratory medicine,Internal medicine,Pediatric hematology
Keywords: Chronic myeloid leukemia, childhood, WHO classification, case report
©Abir Yahyaoui et al. PAMJ Clinical Medicine (ISSN: 2707-2797). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Abir Yahyaoui et al. Pediatric chronic myeloid leukemia: a case report of a disease with a unique biology. PAMJ Clinical Medicine. 2024;14:41. [doi: 10.11604/pamj-cm.2024.14.41.42993]
Available online at: https://www.clinical-medicine.panafrican-med-journal.com//content/article/14/41/full
Case report 

Pediatric chronic myeloid leukemia: a case report of a disease with a unique biology
Pediatric chronic myeloid leukemia: a case report of a disease with a unique biology
![]() Abir Yahyaoui1,2,&, Imane Douichi1,2, Nada Ouahabi1,2, Wissame Azizi1,2, Abdessamad Amrani1,2, Abdelilah Berhili1,2, Nabiha Trougouty1,2, Mounia Slaoui1,2, Mohammed Bensalah1,2, Ayad Ghanam 2,3, Rachid Seddik1,2
Abir Yahyaoui1,2,&, Imane Douichi1,2, Nada Ouahabi1,2, Wissame Azizi1,2, Abdessamad Amrani1,2, Abdelilah Berhili1,2, Nabiha Trougouty1,2, Mounia Slaoui1,2, Mohammed Bensalah1,2, Ayad Ghanam 2,3, Rachid Seddik1,2
&Corresponding author
Chronic myeloid leukemia is a myeloproliferative syndrome due to monoclonal myeloid proliferation without maturation arrest, predominantly in the granular lineage with passage of immature granular elements into the peripheral blood, defined by the existence of a cytogenetic abnormality constantly associated with the disease, the translocation (9;22) translocation, which moves the ABL gene from chromosome 9 to chromosome 22, close to a BCR breakpoint resulting in the formation of the Philadelphia chromosome, which generates a chimeric BCR-ABL1 oncogene. This is a rare and unusual disease in the pediatric population. However, children and adolescents tend to have a more aggressive clinical presentation than adults. We report the case of chronic myeloid leukemia in a nine-year-old child, revealed by prolonged fever, bone pain, and altered general condition, with a karyotype showing the translocation (9;22), and a hyperdiploid subclone with 47 chromosomes and an additional trisomy 8. It´s the youngest patient diagnosed with chronic myeloid leukemia in our institution to date, to clarify the clinical and biological particularities of this entity with a literature review and a classification challenge in the new WHO 2022 edition.

Chronic myeloid leukemia (CML) is a myeloproliferative syndrome caused by monoclonal myeloid proliferation without maturation arrest, predominantly in the granular lineage, with the passage of immature granular elements into the peripheral blood. This is the first pathology directly correlated with a clonal-acquired cytogenetic abnormality. It is a rare disease in children and adolescents, accounting for 2-3% of all leukemias in the pediatric population under the age of 15 [1]. It is more common in adults over 60 years of age, with a median age at diagnosis of 67 years [2]. It is defined by the existence of a cytogenetic anomaly constantly associated with the disease, the translocation (9;22), which moves the ABL gene from chromosome 9 to chromosome 22, close to a BCR breakpoint, resulting in the formation of a shortened chromosome 22, known as the Philadelphia chromosome (Ph). This generates a chimeric BCR-ABL1 oncogene, which encodes a protein with high tyrosine kinase activity, responsible for clonal cell proliferation, inhibition of apoptosis, and genomic instability. Given the rarity of this diagnosis and clinical trial data, current management recommendations are derived from studies or practice guidelines developed for adult CML patients [3]. However, there are clear differences between pediatric and adult CML in terms of clinical presentation, biology, disease course, and side effects that need to be taken into account when treating pediatric CML patients [2]. We report the case of chronic myeloid leukemia in a nine-year-old child, the youngest patient treated for CML in our group, to clarify the clinical and biological particularities of this entity.

Patient information: the child was nine years old, male, from a second-degree consanguineous marriage, and the youngest of four siblings. Admitted to the pediatric emergency department for management of prolonged fever, anemic syndrome, and altered general condition.
Clinical finding: clinical examination revealed a conscious patient, hemodynamically and respiratory stable, with mucocutaneous pallor and tumor syndrome consisting of splenomegaly and bilateral inguinal adenopathy. The patient suffered from anorexia, asthenia, and weight loss.
Timeline of the current episode: the history of the illness dates back to two weeks before admission, with the onset of a fever that was not quantified and increased at night, with nocturnal sweating, accompanied by increasing deterioration in the general condition.
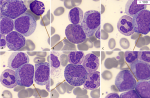
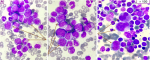
Diagnostic assessment: the complete blood count received at the central laboratory of the hematology department on the day of admission showed hyperleukocytosis at 332890/μL (age reference range [RR]: 4000-14500/μL), normocytic normochromic anemia at 10 g/dl ([RR]: 11.1-14.7 g/dL) and thrombocytosis at 526000/μL (RR: 166000-463000/μL) with a scattergram showing the presence of a grayish cloud corresponding to a large, highly fluorescent population located in the immature granulocyte zone. A blood smear was then taken, after spreading and staining with May-Grünwald Giemsa (MGG), showing neutrophilic polynucleosis with high myelemia (38%) made up of myelocytes, metamyelocytes and promyelocytes, neutrophils and 4% blasts of myeloid appearance, large size, high nucleocytoplasmic ratio, irregular nuclear contour, fine nucleated chromatin and basophilic cytoplasm, as well as 9% eosinophilia and 3% basophilia (Figure 1). Subsequent bone marrow aspiration showed hyperplasia of the neutrophilic granulocytic lineage at all stages of maturation, with promyelocyte, hyper granular myelocyte, metamyelocyte and increased eosinophilic and basophilic lineages with erythroblastopenia. Megakaryocytes were smaller and hypo-segmented, and blasts represented 05% of bone marrow cellularity (Figure 2). The multiparametric biochemical workup showed the LDH level increased to 1162 IU/L, the blood ionogram, uremia, and creatininemia were normal, and the uric acid level was 46mg/l. Hemostasis assays were unremarkable. Cytogenetic analysis of the bone marrow as part of the etiological work-up confirmed the presence of the Philadelphia chromosome with translocation t (9;22) between the long arm of chromosome 9 at q34 and the arm of chromosome 22 at q11. The karyotype showed an initial pseudodiploid clone with 46 chromosomes and a clonal structural abnormality, the translocation (9;22), and a hyperdiploid subclone with 47 chromosomes and an additional trisomy 8, representing a major additional cytogenetic abnormality requiring increased surveillance. Molecular testing for the BCR-ABL1 fusion transcript by RT-PCR on EDTA whole blood detected 50% (IS).
Diagnosis: the diagnosis of chronic myeloid leukemia was retained on the basis of cytological, karyotype and molecular biology criteria established by the WHO, in 2022, and acute leukemia has been eliminated.
Therapeutic interventions: the child was hospitalized and began symptomatic treatment with hyperhydration to prevent lysis syndrome. He was treated with hydroxyurea and then Imatinib, a 1st generation tyrosine kinase inhibitor (TKI), which was started after confirmation of the diagnosis.
Follow-up and outcome of interventions: biological evolution was marked by a decrease in white blood cell count to 62550/μL, persistence of normocytic normochromic anemia, and thrombocytosis to 643000/μL. Molecular evaluation is planned at three months' follow-up.
Patient perspective: the patient and his family were satisfied and grateful for the quality of the care provided to their child, as well as his favorable and spectacular evolution after starting the treatment.
Informed consent: the patient and his parents have expressed their consent to the publication of this report.
Chronic myeloid leukemia (CML) is a rare hematological malignancy in the pediatric population. Annual incidence increases with age from 1 per million in children under 15 to 2.5 per million in adolescents [4]. The clinical and laboratory presentation of pediatric CML differs from that of adults. The median white blood cell (WBC) count at baseline in adult CML ranges from 80 to 150G/L. Analysis of data from a randomized study of CML showed that adolescents and young adults had a clinical feature suggestive of disease, with larger spleens, higher WBC counts, a high percentage of peripheral blasts, and lower-than-normal hemoglobin compared with other age groups [5]. Our patient tested positive for the Philadelphia chromosome, with the presence of an additional cytogenetic abnormality, trisomy 8 and a hyperdiploid subclone with 47 chromosomes. Nevertheless, children with CML share the same genetic features as adults, the balanced translocation (9;22) (q34; q11), which results in fusion of the ABL1 oncogene located on chromosome 9 to the same breakpoint regions in the Major BCR (M-BCR) gene on chromosome 22, leading to constitutive dysregulation of the ABL1 tyrosine kinase. The distribution pattern detected in children is similar to that observed in adult Philadelphia-positive acute lymphoblastic leukemia (Ph1 ALL) with a rearrangement of the M-BCR gene. Recently, a higher incidence of mutated cancer driver genes has been found in children with CML compared with adults with CML [6].
A study of a large pediatric case series showed that in children, as in adults, specific BCR/ABL1 transcript types are associated with distinct hematological alterations, such as elevated platelet counts with the e14a2 transcript [7]. As in adults, three phases of evolution, chronic (CP), accelerated (AP) and blast (BC), can be recognized in childhood CML according to the 2016 World Health Organization (WHO) classification of myeloid hemopathies [8]. Changes have been made in the new 2022 edition, however chronic phase (CP) and blast phase (BP) are identified as essential phases of the disease accelerated phase (AP) is no longer required and the risk factors for chronic myeloid leukemia are refined. Thus, the designation of the accelerated phase has become less relevant, as resistance arising from ABL1 kinase mutations and/or additional cytogenetic abnormalities and the development of the blast phase represent key features of the disease. Furthermore, the presence of lymphoblasts, even in low numbers, may herald blast transformation and require further evaluation [9].
Furthermore, the international consensus classification (ICC) of myeloid neoplasia and acute leukemia, updated in 2022, still considers AP, and defines progression to this phase by the presence of continuous BCR-ABL1 activity, increased genomic instability, and DNA damage. This leads to clonal proliferation with the acquisition of BCR-ABL1 kinase domain mutations and additional cytogenetic abnormalities (ACA), considered characteristic of CML in AP. The ICC has maintained a blast percentage threshold of 10-19% for AP and at least 20% in blood or bone marrow to establish the diagnosis of BP. The presence of an increasing number of lymphoblasts (>5%) in peripheral blood (PB) or bone marrow (BM) leads to the consideration of a lymphoblastic crisis with the need for immunophenotyping to confirm the lymphoid lineage [10]. As for treatment, our patient benefited from specific Imatinib therapy. According to the literature, Imatinib is the first-line drug. However, discontinuation of Imatinib after prolonged complete molecular remission is not yet recommended in children [1]. Our patient responded well to medical treatment based on tyrosine kinase inhibitors. Good therapeutic compliance and rigorous biological follow-up are required in order to improve the management of this rare entity in children.
Pediatric chronic myeloid leukemia is a rare and unusual disease with considerable differences from adults. Recent studies on molecular biology are beginning to elucidate the differences between pediatric and adult, which may explain the differences in clinical and biological presentation and disease course. Thus, one of the major challenges in managing this pathology is to use a stratification score specific to this age group, as well as its classification in the new WHO 2022 edition, to predict progression and adapt therapeutic management.
The authors declare no competing interests.
All authors have read and approved the final of this manuscript.
We would like to thank all the staff of the biological hematology department of our laboratory for their valuable contributions.
Figure 1: (A-F) blood smear of our patient showing significant myeloma with the presence of various precursors of the myeloid lineage, basophilia, eosinophilia, and blasts (MGG stain, x100 objective)
Figure 2: bone marrow smear of our patient showing granular lineage hyperplasia; (A) with a micro-megakaryocyte; (B) (MGG stain, x100 objective)
- Hijiya N, Millot F, Suttorp M. Chronic myeloid leukemia in children: clinical findings, management, and unanswered questions. Pediatr Clin North Am. 2015 Feb;62(1):107-19. PubMed | Google Scholar
- Millot F, Traore P, Guilhot J, Nelken B, Leblanc T, Leverger G et al. Clinical and biological features at diagnosis in 40 children with chronic myeloid leukemia. Pediatrics. 2005 Jul;116(1):140-3. PubMed | Google Scholar
- Pemmaraju N, Kantarjian H, Shan J, Jabbour E, Quintas-Cardama A, Verstovsek S et al. Analysis of outcomes in adolescents and young adults with chronic myelogenous leukemia treated with upfront tyrosine kinase inhibitor therapy. Haematologica. 2012 Jul;97(7):1029-35. PubMed | Google Scholar
- NIH. SEER Cancer Statistics Review, 1975-2017. Accessed November 23, 2023.
- Pushpam D, Bakhshi S. Paediatric chronic myeloid leukaemia: Is it really a different disease. Indian J Med Res. 2019 May;149(5):600-609. PubMed | Google Scholar
- Ernst T, Busch M, Rinke J, Ernst J, Haferlach C, Beck JF et al. Frequent ASXL1 mutations in children and young adults with chronic myeloid leukemia. Leukemia 2018 Sep;32(9):2046-2049. PubMed | Google Scholar
- Adler R, Viehmann S, Kuhlisch E, Martiniak Y, R�ttgers S, Harbott J et al. Correlation of BCR/ABL transcript variants with patients´ characteristics in childhood chronic myeloid leukaemia. Eur J Haematol 2009 Feb;82(2):112-8. PubMed | Google Scholar
- Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016 May 19;127(20):2391-405. PubMed| Google Scholar
- Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022 Jul;36(7):1703-1719. PubMed | Google Scholar
- Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022 Sep 15;140(11):1200-1228. PubMed| Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures
Article metrics

