
Vascularite urticarienne hypocomplémentémique: à propos d´un cas et revue de la littérature
El Houcine El Idrissi, Naoufal Assoufi
Corresponding author: El Houcine El Idrissi, Service de Médecine Interne, Hôpital Militaire Oued Eddahab, Agadir, Maroc 
Received: 20 Jan 2024 - Accepted: 19 Mar 2024 - Published: 24 May 2024
Domain: Internal medicine
Keywords: Urticaire, vascularite, complément, anticorps anti-C1q, cas clinique
©El Houcine El Idrissi et al. PAMJ Clinical Medicine (ISSN: 2707-2797). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: El Houcine El Idrissi et al. Vascularite urticarienne hypocomplémentémique: à propos d´un cas et revue de la littérature. PAMJ Clinical Medicine. 2024;15:9. [doi: 10.11604/pamj-cm.2024.15.9.42731]
Available online at: https://www.clinical-medicine.panafrican-med-journal.com//content/article/15/9/full
Case report 

Vascularite urticarienne hypocomplémentémique: à propos d´un cas et revue de la littérature
Vascularite urticarienne hypocomplémentémique: à propos d'un cas et revue de la littérature
Hypocomplementemic urticarial vasculitis syndrome (HUVS): a literature review and a case study
![]() El Houcine El Idrissi1,&,
El Houcine El Idrissi1,&, ![]() Naoufal Assoufi1
Naoufal Assoufi1
&Auteur correspondant
La vascularite urticarienne hypocomplémentémique (VUH) est une vascularite systémique des petits vaisseaux associant une urticaire récidivante, une hypo-complémentémie et la présence d'anticorps anti-C1q du complément. Nous rapportons un cas de VUH avec des manifestations articulaires, abdominales et oculaires. Nous rapportons le cas d'une patiente âgée de 23 ans, suivie pour un asthme depuis l'âge de 2 ans, admise dans notre service pour des poussées récurrentes d'une urticaire généralisée associée à une polyarthralgie inflammatoire, des douleurs abdominales et des signes de conjonctivite. La biologie avait montré un syndrome inflammatoire, la présence de l'anticorps anti-C1q sans baisse du complément sérique et des anticorps antinucléaires négatifs. La biopsie cutanée a objectivé un dépôt de C1q sans lésions vascularitiques. La patiente a été traitée par colchicine et hydroxychloroquine avec une évolution favorable. La VUH de McDuffie est une affection rare. L'anticorps anti C1q est un élément évocateur de la maladie mais n'est pas spécifique et le complément sérique n'est pas toujours consommé.
Hypocomplementemic Urticarial Vasculitis Syndrome (HUVS) is a small vessel systemic vasculitis characterized by recurrent urticaria, hypocomplementemia and the presence of complement anti-C1q antibodies. We here report the case of a patient with articular, abdominal, and ocular manifestations of HUVS. A 23-year-old female patient with asthma on treatment since the age of 2 was admitted with generalized urticaria, inflammatory polyarthralgia, abdominal pain, and conjunctivitis. Laboratory findings revealed an inflammatory syndrome, the presence of anti-C1q antibodies without a decrease in serum complement levels. Antinuclear antibodies were negative. Skin biopsy showed C1q deposits without vasculitic lesions. The patient was treated with colchicine and hydroxychloroquine, with favorable evolution. McDuffie's syndrome, or HUVS, is a rare condition. Anti-C1q antibodies, though suggestive, are not disease-specific, and serum complement levels may not always be depleted.
Key words: Urticaria, vasculitis, complement, anti-C1q antibody

La vascularite urticarienne hypo-complémentémique (VUH), est une maladie rare qui associe une vascularite urticarienne cutanée, une hypocomplémentémie et des atteintes systémiques. Elle est également connue sous les noms vascularite avec anticorps anti-C1q ou syndrome de McDuffie. La VUH peut être associée à d'autre maladie systémique telle que le lupus érythémateux systémique (LES), le Syndrome de Gougerot-Sjögren (SGS), la vascularite associée aux anticorps du cytoplasme des polynucléaires neutrophiles (ANCA) ou une hémopathie. Le terme de VUH a été retenu dans la conférence de consensus internationale révisée de 2012 de Chapel Hill [1]. Les critères diagnostiques ont été proposés en 1982 par Schwartz et al. [2]. L'association d'une urticaire chronique de plus de six mois, d'une hypocomplémentémie, et la présence de deux critères parmi les suivants sont nécessaires pour retenir le diagnostic (arthralgie/arthrite, angio-oedème, glomérulonéphrite (GN), atteinte oculaire inflammatoire, atteinte digestive, vascularite leucocytoclasique et présence d'anticorps (Ac) anti-C1q).

Nous rapportons un cas d'une patiente présentant le syndrome de McDuffie avec des manifestations articulaires, digestives et oculaires.
Information de la patiente
Notre observation concerne une patiente de 23 ans, ayant comme antécédent: un asthme depuis l'âge de 2 ans bien contrôlé sous bêta-2-mimétique, une amygdalectomie depuis un an devant des angines à répétitions. La malade a été hospitalisée au service de médecine interne au mois de juillet 2023 pour bilan étiologique d'une urticaire chronique associée à des arthralgies inflammatoires et des douleurs abdominales.
Le début de la symptomatologie remonte à 2009 par l'apparition d'une éruption cutanée maculo-papuleuse en placard touchant le tronc et les membres associée à une polyarthralgie inflammatoire des moyennes et des grosses articulations. Les lésions cutanées avaient comme caractéristique: persistante de 12 à 24 heures, apparition surtout le soir, prurit intense, accompagnée des douleurs lors des poussées cutanées (des algies de la paume des mains et la plante du pied). L'évolution de l'éruption est par poussée et ces lésions urticariennes dépassent parfois les 24h sans laisser des traces. La patiente rapportait aussi deux épisodes d'angio-œdème du visage, un en 2015 et un autre en 2021. De plus, la malade présentait une photosensibilité marquée avec des brûlures oculaires. Sur le plan respiratoire, elle rapportait une dyspnée stade I de Sadoul depuis 2 ans sans autres signes respiratoires (pas de toux, ni expectoration, ni hémoptysie, ni sifflement thoracique). Le tout évoluant dans un contexte d'apyrexie et de conservation de l'état général. Nous avons noté la persistance des lésions urticariennes malgré la prise de plusieurs antihistaminiques.
Résultats cliniques
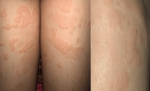
À l'admission, la patiente était apyrétique à 36,5°C, normotendue à 11/7 cmHg, présentant une éruption cutanée maculo-papuleuse érythémateuse en placard, une rougeur oculaire bilatérale, des arthralgies inflammatoires touchant les coudes, poignets et les genoux. La patiente avait des douleurs abdominales des deux flancs plus intenses lors des poussées cutanées associées à des nausées. L'examen clinique trouvait des lésions d'urticaire en placard sur le tronc, les cuisses, les bras et les mains (Figure 1). Les articulations sont libres sans signes d'arthrite. L'examen abdominal était normal. L'examen ophtalmologique objectivait des signes de conjonctivite. L'étude des sécrétions lacrymales était sans anomalie.
Démarche diagnostique
Sur le plan biologique, il existait un discret syndrome inflammatoire avec une vitesse de sédimentation (VS) à 29 mm à la première heure, une CRP à 13,2 mg/l. L'hémoglobine était à 12,6 g/dl (pas d'hyperéosinophilie ni de lymphopénie). Les sérologies de l'hépatite B et C, HIV et la protéinurie de 24 h étaient négatives. Le bilan immunologique objectivait l'anticorps anti C1q + à 33 U/ml (> à 10 Ui / ml). Le dosage du complément (C3, C4, CH50) était dans les normes. Le dosage pondéral du C1 inhibiteur était normal à 0,38 g/L (N: 0,21- 0,39). La recherche d'anticorps (Ac) antinucléaires, Ac anti-DNA natifs, ANCA, antistreptolysines O (ASLO) et de facteur rhumatoïde était négative. Le profil de l'électrophorèse des protéines plasmatiques était normal.
Le scanner thoracique et l'exploration fonctionnelle respiratoire étaient normaux (une capacité pulmonaire totale (CPT) à 4,66 (93%), diffusion de monoxyde de carbone (CO) à 8,56 (91%)). L'échographie abdominale était normale et l'exploration cardiaque était sans particularité. La biopsie cutanée retrouvait un discret remaniement fibreux, l'immunofluorescence directe a montré des dépôts linéaires sous épidermique de C1q. La biopsie de glandes salivaires n'a pas retrouvé d'infiltration lympho-plasmocytaire. Le diagnostic de vascularite urticarienne a été retenu devant l'urticaire chronique, les arthralgies inflammatoires, les manifestations digestives, la présence d'Ac anti-C1q et le dépôt du complément C1q au niveau cutané.
Intervention thérapeutique et suivi
La patiente a été mise sous colchicine associée à l'hydroxychloroquine 400 mg/j. L'évolution était favorable en quelques jours, avec la disparition des poussées d'urticaire, l'amélioration des arthralgies et de la dyspnée. Cependant, nous avons noté la persistance de douleurs abdominales.
Perspectives de la patiente
« Quand j'ai commencé le traitement, les symptômes comme les démangeaisons, la rougeur, l'inflammation de la peau et la difficulté à respirer ont disparu carrément. Mais les douleurs abdominales sont présentes la plupart du temps. À propos des médicaments, ils m'ont causé des vertiges pendant les premiers jours seulement ».
Consentement éclairé: le consentement éclairé de la patiente a été obtenu.
La vascularite urticarienne hypocomplémentémique (VUH) est une vascularite systémique rare, rapportée pour la première fois par McDuffie en 1973 [2]. Elle touche le plus souvent les adultes de 30 à 50 ans, avec une prédominance féminine [3]. Elle est caractérisée par une activation du complément, une diminution du C1q et la présence de C1q précipitines [4]. L'atteinte cutanée est constante, caractérisée par une éruption urticarienne le plus souvent fixe et persistante (parfois plus de 24 heures). La maladie peut évoluer et donner des atteintes systémiques, notamment articulaires, abdominales et pulmonaires [5,6].
La physiopathologie de la VUH serait médiée par les complexes immuns. La formation de ce complexe et leur dépôt dans les parois vasculaires vont aboutir à l'activation du complément. Les anticorps anti-C1q peuvent jouer un rôle dans la formation du complexe immun. L'activation du complément génère du C3a et du C5a, qui activent la dégranulation des mastocytes, ce qui entraîne des lésions urticariennes au niveau de la peau. Ainsi, les vascularites urticariennes sont considérées comme une réaction d'hypersensibilité de type 3 et ces réactions favorisent l'œdème et l'inflammation des parois vasculaires [6,7].
Sur le plan dermatologique, les lésions urticariennes peuvent être douloureuses ou prurigineuses. Elles prédominent aux membres et au tronc, et peuvent toucher la face et parfois les paumes et les plantes. L'angio-œdème est présent dans environ 50-72% des cas [6,8]. La biopsie cutanée révèle souvent une vascularite leucocytoclasique. L'immunofluorescence directe (IFD) est positive dans environ la moitié des cas, montrant des dépôts d'immunoglobulines et du complément (C3, C4, C1q). Parfois, la vascularite histologique peut être méconnue si la biopsie porte sur des lésions en voie de guérison [6,7]. Comme dans notre cas, l'étude histologique n'a pas objectivé de vascularite cutanée, en revanche un dépôt de complément (C1q) a été retrouvé sur les biopsies cutanées.
Les manifestations articulaires sont assez fréquentes à type d'arthralgie ou d'arthrite. La distribution est souvent oligo- ou polyarticulaire, touchant les grosses et moyennes articulations. L'atteinte articulaire est non déformante sauf dans le cas d'arthropathie de Jaccoud (décrite rarement dans le cadre du VUH) [5,7]. Chez notre patiente, la polyarthralgie a été considérée initialement une des manifestations de rhumatisme articulaire aigü. Mais après l'amygdalectomie, les algies articulaires sont persistantes.
Les douleurs abdominales sont décrites dans environ 40% des cas. L'atteinte digestive se manifeste principalement par des douleurs abdominales, des diarrhées, des nausées et des vomissements. Ces manifestations surviennent simultanément aux poussées d'urticaire [3]. Quelques cas d'ascite ont été rapportés, ainsi que des cas de pancréatite aiguë et une localisation de vascularite digestive dans la vésicule biliaire [5]. Dans notre observation, les douleurs abdominales sont encore inexpliquées et persistent malgré le traitement médical.
L'atteinte pulmonaire est parfois caractéristique de la maladie. Il s'agit d'un syndrome obstructif avec emphysème révélé le plus souvent par la toux et/ou une dyspnée [5,6]. L'atteinte obstructive est trouvée dans 20 à 50% des cas et peut simuler un asthme [6]. Chez notre malade, l'asthme remonte à l'âge de 2 ans et son association avec la VUH est difficile à approuver vue que l'asthme précède le début des poussées cutanées avec un intervalle de 7 ans. Concernant l'atteinte oculaire, elle est signalée dans 32% des cas. Il s'agit principalement d'uvéites, d'épisclérites ou de conjonctivites [3]. L'atteinte rénale est observée dans 25% des cas. La protéinurie en est le signe d'appel le plus fréquent. L'insuffisance rénale est une complication rare. Le reste des manifestations (cardiaque, neurologique, ganglionnaire, musculaire) sont plutôt rares [5,9].
Dans notre observation, nous avons un tableau clinique regroupant des manifestations cutanées, articulaires, abdominales et oculaires. L'atteinte systémique était évocatrice de la VUH surtout avec un titre élevé de l'anticorps anti C1q, un dépôt de complément type C1q dans les lésions cutanées et l'absence des éléments en faveur d'autres pathologies notamment le lupus systémique (LES). Un dosage du complément sérique était normal dans notre observation, ce qui suggère de répéter ce dosage lors des poussées antérieures de la maladie. La plupart des études objectivaient une baisse du complément dans presque 100% des cas avec une baisse de C1q [5,10].
Sur le plan clinique, la VUH partage de nombreuses caractéristiques communes avec le lupus systémique [8]. La confusion est donc possible d'autant plus que l'anticorps anti-C1q n'est pas spécifique de McDuffie. La présence de cet anticorps dans le LES est estimée de 28 à 47% des cas. L'élément principal de différence est l'absence des anticorps antinucléaires et de l'anti-DNA dans la VUH. Un chevauchement est aussi possible entre ces deux pathologies ou avec d'autres maladies auto-immunes notamment le syndrome de Sjögren [5].
Selon la littérature, les antihistaminiques ne sont pas efficaces sur l'atteinte cutanée du VUH. Ainsi, le traitement classique repose sur d'autres molécules. Les classes thérapeutiques de première ligne sont représentées par hydroxychloroquine (HCQ), colchicine ou dapsone. Ces trois classes sont aussi efficaces qu'une corticothérapie [5,8]. La décision thérapeutique chez notre patiente était d'associer la colchcine avec hydroxychloroquine pendant 3 mois puis de laisser la malade sous HCQ seule. L'efficacité thérapeutique a été observée pendant un recul de 6 mois.
La vascularite urticarienne hypocomplémentémique est une entité rare, caractérisée par des signes cliniques allant des signes cutanés isolés à des manifestations systémiques. L'anticorps anti-C1q est un élément évocateur de la maladie mais n'est pas spécifique et l'hypocomplémentémie n'est pas toujours présente.
Les auteurs ne déclarent aucun conflit d'intérêts.
Collecte, analyse, interprétation des données et rédaction de l'article: El Houcine El Idrissi. Révision de l'article: Naoufal Assoufi. Tous les auteurs ont approuvé la version finale du manuscrit.
Figure 1: multiple lésions d'urticaires sur les deux cuisses
- Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013 Jan;65(1):1-11. PubMed | Google Scholar
- Schwartz H, McDuffie F, Black L, Schroeter A, Conn D. Hypocomplementemic urticarial vasculitis: association with chronic obstructive pulmonary disease. Mayo Clin Proc. 1982 Apr;57(4):231-8. PubMed | Google Scholar
- el Maghraoui A, Abouzahir A, Mahassine F, Tabache F, Bezza A, Ghafir D et al. Vascularite urticarienne hypocomplémentémique de McDuffie. Deux observations et revue de la littérature. Rev Med Interne. 2001 Jan;22(1):70-4. PubMed | Google Scholar
- Zeiss CR, Burch FX, Marder RJ, Furey NL, Schmid FR, Gewurz H. A hypocomplementemic vasculitic urticarial syndrome. Report of four new cases and definition of the disease. Am J Med. 1980 Jun;68(6):867-75. PubMed | Google Scholar
- Jachiet M, Flageul B, Bouaziz J, Bagot M, Terrier B; Groupe Fran�ais d'Étude des Vascularites (GFEV). Les vascularites urticariennes hypocomplémentémiques. Rev Med Interne. 2018 Feb;39(2):90-98. PubMed | Google Scholar
- Jara LJ, Navarro C, Medina G, Vera-Lastra O, Saavedra MA. Hypocomplementemic urticarial vasculitis syndrome. Curr Rheumatol Rep. 2009 Dec;11(6):410-5. PubMed | Google Scholar
- Hamad A, Jithpratuck W, Krishnaswamy G. Urticarial vasculitis and associated disorders. Ann Allergy Asthma Immunol. 2017 Apr;118(4):394-398. PubMed | Google Scholar
- Buck A, Christensen J, McCarty M. Hypocomplementemic urticarial vasculitis syndrome: a case report and literature review. J Clin Aesthet Dermatol. 2012 Jan;5(1):36-46. PubMed | Google Scholar
- Messiaen T, Van Damme B, Kuypers D, Maes B, Vanrenterghem Y. Crescentic glomerulonephritis complicating the course of a hypocomplementemic urticarial vasculitis. Clin Nephrol. 2000 Nov;54(5):409-12. PubMed | Google Scholar
- Wisnieski JJ, Baer AN, Christensen J, Cupps TR, Flagg DN, Jones JV et al. Hypocomplementemic urticarial vasculitis syndrome. Clinical and serologic findings in 18 patients. Medicine (Baltimore). 1995 Jan;74(1):24-41. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures
Article metrics

