
Myofibrome infantil solitaitre: à propos d’un cas
Fatima-Zahra Agharbi
Corresponding author: Fatima-Zahra Agharbi, CHR Civil Tétouan,Tétouan, Maroc 
Received: 16 Dec 2019 - Accepted: 11 Feb 2020 - Published: 14 Feb 2020
Domain: Dermatology
Keywords: Myofibrome, myofibromatose, localisée, multiple, généralisée
©Fatima-Zahra Agharbi et al. PAMJ Clinical Medicine (ISSN: 2707-2797). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Fatima-Zahra Agharbi et al. Myofibrome infantil solitaitre: à propos d’un cas. PAMJ Clinical Medicine. 2020;2:51. [doi: 10.11604/pamj-cm.2020.2.51.21323]
Available online at: https://www.clinical-medicine.panafrican-med-journal.com//content/article/2/51/full
Case report 
Myofibrome infantil solitaitre: à propos d’un cas
Myofibrome infantile solitaire: à propos d´un cas
Myofibroma infantil solitary: about a case
Fatima-Zahra Agharbi1,&
1CHR Civil Tétouan,Tétouan, Maroc
&Auteur correspondant
Fatima-Zahra Agharbi, CHR Civil Tétouan,Tétouan, Maroc
La myofibromatose infantile (MFI) décrite par Enzinger en 1981 est la plus fréquente des fibromatoses de l´enfant. Elle est constituée par une prolifération bénigne de fibroblastes et de myofibroblastes. Il en existe trois formes. La MFI solitaire, la plus fréquente, caractérisée par une lésion isolée cutanée, osseuse ou des tissus mous; la MFI multiple représentée par plusieurs localisations et la MFI généralisée comportant une atteinte viscérale. Les caractères morphologiques sont identiques quelle que soit la localisation. Le diagnostic histologique repose sur l´identification d´une double composante, myofibroblastique fasciculée en périphérie et d´aspect hémangiopéricytaire au centre qui exprime l´actine musculaire lisse. Il n´existe le plus souvent pas d´atypie, ni de mitose, mais il peut être observé des images d´extensions intravasculaires au centre. Dans la forme solitaire, le pronostic de la MFI est bon mais il est souvent fatal en cas d´atteinte viscérale. L´observation rapportée concerne un enfant de 4 ans qui présente depuis l´âge d'un an un nodule inguinal gauche dont l´étude histologique après exérèse chirurgicale a permis de retenir le diagnostic de myofibrome.
Infantile myofibromatosis (ILM) described by Enzinger in 1981 is the most common childhood fibromatosis. It consists of a benign proliferation of fibroblasts and myofibroblasts. There are three forms. Solitary MFI, the most common type, characterized by isolated skin, bone or soft tissue injury; multiple MFI represented by several locations and generalized MFI with visceral involvement. The morphological characters are identical whatever the location. The histological diagnosis is based on the identification of a double component, myofibroblastic fasciculated at the periphery and hemangiopericy at the center, which expresses smooth muscle actin. There is usually no atypia or mitosis, but there can be observed images of intravascular extensions in the center. In the solitary form, the prognosis of MFI is good but it is often fatal in cases of visceral involvement. The reported case concerns a 4-year-old child who has had a left inguinal nodule since the age of 1, whose histological study after surgical excision has led to the diagnosis of myofibroma.
Key words: Myofibroma, myofibromatosis, localized, multiple, generalised

La myofibromatose infantile est la plus fréquente des fibromatoses de l´enfant. Elle est constituée par une prolifération bénigne de fibroblastes et de myofibroblastes. Il en existe trois formes: La forme solitaire, la forme multiple et la forme généralisée. Le pronostic de MFI est généralement bon en dehors de la forme généralisée comportant une atteinte viscérale.

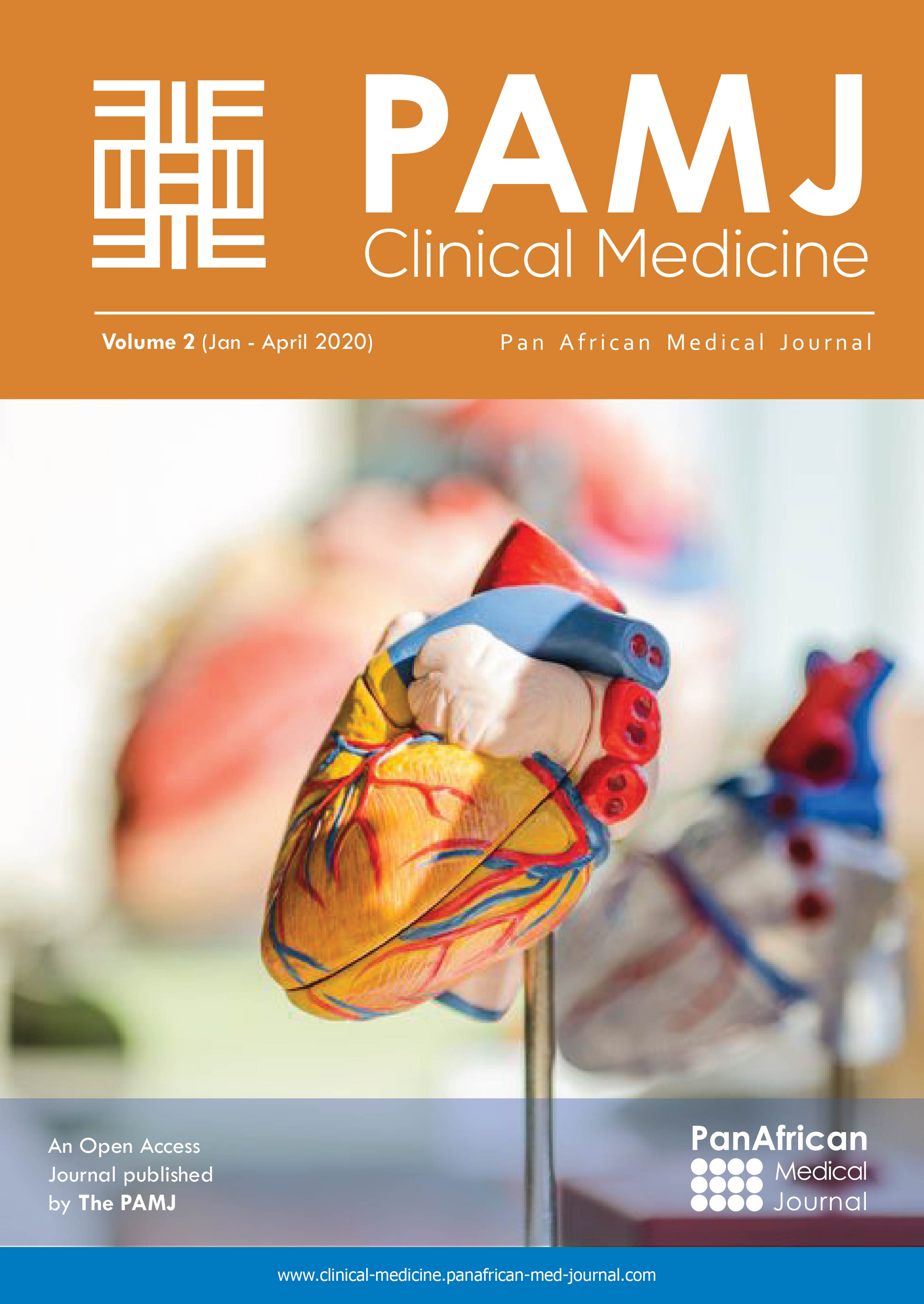
Enfant de 4 ans, présentant depuis l´âge d´un an un nodule inguinal gauche augmentant progressivement de volume asymptomatique. L´examen clinique trouvait un nodule de 1 cm à surface violacée siégeant au niveau inguinal gauche (Figure 1) dont l´étude histologique après exérèse était en faveur d´un myofibrome.
Les myofibromes sont des tumeurs des tissus mous dont les caractéristiques varient en fonction de l´âge auquel ils sont découverts. La myofibromatose a été rapportée pour la première fois chez l´enfant par Stout en 1954 et appelée « fibromatose congénitale généralisée ». Divers synonymes existent dans la littérature (hamartomatose généralisée, hamartomes mésenchymateux congénitaux multiples, léiomyomes vasculaires multiples du nouveau-né, fibromatose congénitale multiple) [1]. Le terme de « myofibromatose infantile » a été proposé en 1981 par Chung et Enzinger [2]. On en distingue 2 types, une forme solitaire et une forme disséminée avec de multiples lésions. Cette seconde forme, moins fréquente, peut s´accompagner d´atteinte viscérale dont l´issue est parfois fatale [3]. Chez l´enfant, Les lésions sont le plus souvent présentes à la naissance ou apparaissent au cours des 2 premières années de vie. La forme solitaire est un nodule, rénitent, bien circonscrit et parfois polylobé, de taille variable. Il peut être rouge ou violacé, quelquefois recouvert de télangiectasies [4]. Le siège est ubiquitaire mais le myofibrome prédomine sur la tête et le cou y compris la cavité buccale [3, 4]. Le sexe ratio de 2/1 montre une prédominance masculine [1,3]. Pour ces formes solitaires de l´enfant, il existe une tendance naturelle à la régression spontanée [4]. Dans les myofibromatoses disséminées, les myofibromes sont identiques à ceux des formes solitaires et leurs nombre varient de quelques-uns à plusieurs centaines. Cette forme plus rare se caractérise par une atteinte musculaire, osseuse et viscérale plus fréquente. Le crâne et les os longs seraient plus touchés que le rachis. Les formes limitées à la peau, aux tissus mous et à l´os sont de bon pronostic avec une régression spontanée dans la plupart des cas [2].
L´envahissement viscéral existerait dans 35 p. 100 des formes disséminées. Le décès survient à la naissance ou peu de temps après, par complications cardiaques, pulmonaires ou gastro-intestinales. La mortalité est de l´ordre de 15 p. 100 [1]. Cette gravité potentielle de la forme disséminée justifie la réalisation d´examens complémentaires. Une radiographie du squelette, un scanner corps entier, un ECG et un échocardiogramme sont systématiquement demandés. L´IRM serait plus performante, capable de détecter une involution de la maladie [3]. Une transmission autosomique dominante à pénétrance variable a été proposée en raison de cas familliaux [1, 3, 4]. L´examen morphologique et immunohistochimique montre un nodule bien limité, dermique profond et hypodermique, constitué par une prolifération cellulaire d´aspect biphasique avec un phénomène de zone. En périphérie, on observe une prolifération de cellules fusiformes à cytoplasme abondant éosinophile disposées en faisceaux, de différenciation myoïde. Parfois, ces cellules forment des bourgeons dans les lumières vasculaires dont elles restent séparées par le revêtement endothélial. Au centre, les cellules fusiformes de plus petite taille s´agencent en faisceaux de disposition concentrique autour de vaisseaux ramifiés réalisant un aspect hémangiopéricytaire. Les deux composantes cellulaires expriment également l´alpha-AML et la vimentine et sont négatives pour la desmine et la protéine S100. Cette zone centrale peut être nécrotique, hémorragique ou calcifiée [5,6]. Le choix thérapeutique dépend du caractère solitaire ou multifocal du ou des myofibromes. Le myofibrome solitaire de petite taille est la plupart du temps excisé dans un but diagnostique. Cette exérèse est souvent curatrice. Une récurrence survient dans 10 p. 100 des cas environ [2] et la réexcision est préconisée [3,7]. Pour les autres formes de myofibromatoses décrites chez l´enfant, la possibilité d´une régression spontanée doit toujours être prise en considération, surtout en période néonatale. Le diagnostic sera confirmé par une biopsie.
Lorsque la lésion est unique mais localement agressive ou étendue, la destruction progressive des tissus voisins par la fibrose est susceptible d´entraîner des séquelles fonctionnelles. Certains auteurs préconisent alors une résection partielle afin de préserver les fonctions vitales. L´exérèse complète peut être retardée, dans l´hypothèse d´une involution spontanée de la lésion [8, 9]. Certains myofibromes asymptomatiques pour lesquels un geste chirurgical entraînerait des séquelles esthétiques ou fonctionnelles désastreuses, ne justifient aucun geste thérapeutique. Une surveillance clinique et des examens d´imagerie réguliers sont proposés [10]. Dans les myofibromatoses infantiles disséminées, l´abstention thérapeutique est de mise. La régression spontanée est observée avant l´âge de 2 ans dans la grande majorité des myofibromatoses congénitales se limitant à la peau, aux tissus mous et à l´os. Le mauvais pronostic des formes viscérales est lié à l´effet de masse local des nodules, responsable d´obstruction et de destruction tissulaire [9]. La radiothérapie et la chimiothérapie ont été utilisées pour ces formes non résécables. Les produits utilisés sont la vincristine, l´actinomycine D et le cyclophosphamide, mais l´efficacité est incertaine [9, 11]. Toutefois, en l´absence de retentissement sur les fonctions vitales et malgré une atteinte viscérale diffuse, une régression spontanée est possible après quelques mois [1].
La MFI, bien que la plus fréquente des fibromatoses du nourrisson et de l´enfant, en particulier dans sa forme solitaire et cutanée, constitue une forme rare de lésion mésenchymateuse bénigne de l´enfant. Son diagnostic histologique, dans les formes typiques comme dans notre observation ou dans les formes moins différenciées, repose sur la mise en évidence, en immunohistochimie, d´une différenciation myofibroblastique. Le traitement de choix consiste, dans les formes localisées, en une exérèse chirurgicale, cette affection étant d´excellent pronostic avec dans certains cas, une tendance à la régression spontanée.
Les auteurs ne déclarent aucun conflit d´intérêts.
Tous les auteurs ont lu et approuvé la version finale du manuscrit.
Figure 1: nodule violacé inguinal gauche
- Giannakopoulou C, Hatzidaki E, Giannakopoulos K, Matalliotakis I, Koumantakis E, Kalmanti M. Infantile myofibromatosis: a case study and review of literature. J Dermatol. 1999;26(9):595-8. PubMed | Google Scholar
- Chung EB, Enzinger FM. Infantile myofibromatosis. Cancer. 1981;48(8):1807-18. PubMed | Google Scholar
- Weiss SW, Goldblum JR. Enzinger and Weiss´s Soft tissue tumors. 4th ed St Louis: Mosby. 2001:357-60. Google Scholar
- Stanford D, Rogers M. Dermatological presentations of infantile myofibromatosis: a review of 27 cases. Australas J Dermatol. 2000;41(3):156-61. PubMed | Google Scholar
- Hasegawa T, Hirose T, Seki K, Hizawa K, Okada J, Nakanishi H. Solitary infantile myofibromatosis of bone. Am J Surg Pathol. 1993;17(3): 308-13. PubMed | Google Scholar
- Iijima S, Suzuki R, Otsuka F. Solitary form of infantile myofibromatosis: a histologic, immunohistochemical, and electronmicroscopic study of a regressing tumor over a 20-month period. Am J Dermatopathol. 1999;21(4):375-80. PubMed | Google Scholar
- Hogan SF, Salassa JR. Recurrent adult myofibromatosis: a case report. Am J Clin Pathol. 1992;97(6):810-14. PubMed | Google Scholar
- Netscher DT. Infantile myofibromatosis: case report of a solitary hand lesion with emphasis on differential diagnosis and management. Ann Plast Surg. 2001;46 (1):62-7. PubMed | Google Scholar
- Goldberg NS, Bauer BS, Kraus H, Crussi FG, Esterly NB. Infantile myofibromatosis: a review of clinicopathology with perspectives on new treatment choices. Pediatr Dermatol. 1988;5(1):37-46. PubMed | Google Scholar
- Loundon N, Dedieuleveult T, Ayache D, Roger G, Josset P, Garabedian EN. Head and neck infantile myofibromatosis: a report of three cases. Int J Pediatr Otorhinolaryngol. 1999;51(3):181-6. PubMed | Google Scholar
- Raney B, Evans A, Granowetter L, Schnaufer L, Uri A, Littman P. Nonsurgical management of children with recurrent or unresectable fibromatosis. Pediatrics. 1987;79(3):394-8. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures
Article metrics
PlumX Metrics
Myofibrome infantil solitaitre: à propos d’un cas
