
Rhabdoid tumor of the thyroid gland: a variant of anaplastic carcinoma, case report with review of the literature
Amal Douida, Layla Tahiri, Mohamed Nour-Dine Elalami Elamine, Hind Elfatemi, Laila Chbani, Nawal Hammas
Corresponding author: Amal Douida, Department of Pathology, CHU Hassan II Fes, Fes, Morocco 
Received: 04 Jan 2020 - Accepted: 13 Feb 2020 - Published: 23 Feb 2020
Domain: Oncology
Keywords: Thyroidectomy, anaplastic thyroid carcinoma, rhabdoid cells, immunohistochemistry
©Amal Douida et al. PAMJ Clinical Medicine (ISSN: 2707-2797). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Amal Douida et al. Rhabdoid tumor of the thyroid gland: a variant of anaplastic carcinoma, case report with review of the literature. PAMJ Clinical Medicine. 2020;2:61. [doi: 10.11604/pamj-cm.2020.2.61.21463]
Available online at: https://www.clinical-medicine.panafrican-med-journal.com//content/article/2/61/full
Case report 
Rhabdoid tumor of the thyroid gland: a variant of anaplastic carcinoma, case report with review of the literature
Rhabdoid tumor of the thyroid gland: a variant of anaplastic carcinoma, case report with review of the literature
Amal Douida1,&, Layla Tahiri1,2, Mohamed Nour-Dine Elalami Elamine3, Hind Elfatemi1,2, Laila Chbani1,2, Nawal Hammas1,2
1Department of Pathology, CHU Hassan II Fes, Fes, Morocco, 2Biomedical and Translational Research Laboratory, Medical School of Fez, Sidi Mohamed Ben Abdellah University, Fez, Morocco, 3Department of Oto-Rhino-Laryngology, University Hospital Hassan II, Fez, Morocco
&Corresponding author
Amal Douida, Department of Pathology, CHU Hassan II Fes, Fes, Morocco
Anaplastic thyroid carcinoma (ATC) with rhabdoid features is extremely rare and only few cases have been reported in literature. We report a case of ATC with rhabdoid features in a 66-year-old female, who presented with a rapidly enlarging neck mass. Computed tomography (CT) revealed a multiheteronodular goiter with presence of a suspected nodule in the left lobe. The parient underwent total thyroidectomy with lymph node dissection. Pathology report showed rhabdoid phenotype of thyroid anaplastic carcinoma. This entity is highly aggressive and associated with a poor prognosis. The patient died one month after the operation before receiving adjuvant therapy.

Rhabdoid tumor was first described in 1978 as a variant of Wilms' tumor in children [1] with unfavorable outcome, but designated as a distinct entity in 1981 [2]. Later, this tumor was also reported in the central nervous system [3] and in many other extrarenal sites. Anaplastic thyroid carcinoma (ATC) with rhabdoid features is extremely rare [4] and only few cases have been reported in literature. The rhabdoid phenotype is a pathological presentation associated with aggressive nature not only in thyroid gland but also in other organs [5]. Morphologic findings in this tumor include the presence of large pleomorphic cells with abundant cytoplasm, typical eosinophilic inclusions and eccentric nuclei with distinct nucleoli. We report a case of a rhabdoid variant of a thyroid anaplastic carcinoma and we describe morphologic and immunohistochrmical features of this very rare variant, diagnosis difficulties, prognosis and treatment.

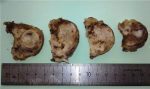
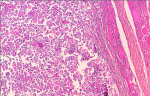
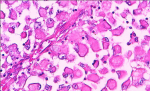
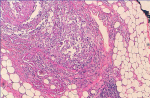
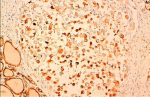
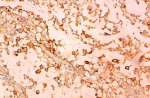
A 66-year-old female presented with painful anterior cervical mass, associated with dyspnea and dysphagia. There was no family history of thyroid disease or radiation exposure to head and neck region. Physical examination revealed an anterior, hard, mobile cervical mass measuring 10cm, without pain or inflammatory signs. Serum thyroid hormones and calcitonin hormone were normal. Cervical ultrasonography showed the presence of multiheterodular goitre with presence of nodules classified TIRADS 4b with adenopathies. Computed tomography (CT) revealed a multiheteronodular goiter with presence of a suspected nodule in the left lobe associated with cervical lymphadenopathies and lung metastatic lesions. Total thyroidectomy, with lymph node dissection were performed. Macroscopic examination revealed in the left lobe a heterogenous nodule measuring 6cm with whitish and blackish areas (Figure 1). Histologically, the tumor was composed of poorly differentiated large cells having an abundant cytoplasm with vitreous eosinophilic inclusion, and eccentric nuclei containing distinct nucleoli (rhabdoid cells) (Figure 2,Figure 3). Vascular emboli was present. This proliferation infiltrated the perithyroid fat tissue (Figure 4). On immunohistochemistry, tumor cells were positive for CKAE1/AE3, EMA and vimentin (Figure 5,Figure 6,Figure 7) and negative for TTF-1, thyroglobulin, chromogranin and synaptophysin. Based on those characteristics, the diagnosis of anaplastic carcinoma in its rhabdoid variant of the thyroid was retained. The patient died one month after the operation before receiving adjuvant therapy.
The initial concept of the rhabdoid cell has been used for the first time in 1978 to describe a variant of Wilms´ tumor in children with a rhabdomyosarcomatous pattern [1]. Eight years later, rhabdoid tumors have been described in different organs in adulte [6-10]. These cells have been observed in extra-renal location and they were not considered as a separate entity but as a final pathway of dedifferentiation of tumors derived from multiple cell types including tumors of neuroectodermal, myogenic, histiocytic, melanocytic and epithelial origin [7,8]. Wick et al. [7] propose to separate these extra-renal tumors into two categories: the first contains only rhabdoids cells and the second contains rhabdoid cells that are combined with another morphological entity, called composite extrarenal rhabdoid tumor. It is interesting to identify the percentage of rhabdoid cells in a tumor, because these cells confer a more aggressive behavior to the tumor. A cutoff of at least 10% of the tumor cell population expressing the rhabdoid phenotype has been proposed as having an adverse prognosis [6].
ATC with rhabdoid features is extremely rare [4] and only few cases have been reported in literature (Table 1). Among these cases, some showed follicular differentiation in the more differentiated areas, while others showed papillary areas, indicating that the rhabdoid differentiation was a part of either follicular or papillary carcinoma of thyroid. In 1991, Chetty and Govender identified this entity in a poorly differentiated follicular thyroid carcinoma [11]. A recent literature review including 12 cases of rhabdoid tumors of the thyroid has shown that the rhabdoid phenotype is reported in 5 cases of follicular carcinoma, 3 cases of papillary carcinoma and 4 cases of anaplastic carcinoma [12]. The case under discussion showed only areas of undifferentiated carcinoma similar to the cases reported by Carmen Carda [13]. ATC can occur at any age [7]. More than half of cases of ATC with rhabdoid features reported in the literature were women with an age ranged from 22 to 77 years with a mean of 57.4 years (Table 1). Clinically, most of patients present with a rapidly enlarging neck mass with local compressive symptoms, like our patient [6].
This variant appears macroscopically as a solid, irregular mass, with a whitish or greyish color [12]. Microscopically, features include the presence of large pleomorphic cells with abundant cytoplasm and characteristic cytoplasmic eosinophilic inclusions and eccentric nuclei [4]. These rhabdoid inclusions should be distinguished from thyroglobulin inclusions observed in follicular neoplasms and containing thyroglobulin [14]. Unlike rhabdoid inclusions, they are periodic acid-Schiff, alcian blue positive and immunoreactive for thyroglobulin. Immunohistochemically, rhabdoid cells tumor of the thyroid gland shows positivity for vimentine and cytokeratins with variable expression of other markers like as TTF-1, EMA, AML, myoglobine, desmin, NSE and PS100 [15]. The rhabdoid tumor cells of the present case are immune-reactive to vimentin, EMA and CK and negative for TTF1, thyroglobulin, chromogranin and synaptophysin. Immunoexpression of SMARCB1 (INI1) in primary childhood malignant rhabdoid tumors is characteristically lost, due to biallelic deletions or mutations in INI1 on chromosome 22 [16].
In adults, the immunoexpression of INI1 helps in distinguishing composite rhabdoid tumors where a rhabdoid cell component, pathogenetically related to primary childhood malignant rhabdoid tumor, coexists with a more differentiated malignancy and shows loss of INI1 expression, from a poorly differentiated carcinoma with rhabdoid phenotype, in which the protein is expressed [5]. The differential diagnosis of such tumor is broad and includes medullary carcinoma of the thyroid, metastatic medullary carcinoma of the kidney, melanomas and variety of sarcomas. These diagnosis are eliminated by the morphological aspect and the immunohistochemical study. The treatment of rhabdoid variant of ATC is not standardized because of the rarity of this entity. Feasible options include surgery, radiotherapy, radio-iodine therapy and chemotherapy [5,11,15,17]. This entity is highly aggressive and associated with a poor prognosis due to lack of response to radio- and chemotherapy. It is associated with local recurrences and metastases with a median survival of 6 months [4]. Some reported thyroid carcinoma with rhabdoid phenotype in the literature are grouped in Table 1 [4,5,11-15,17-20].
Thyroid tumor with rhabdoid phenotype is a rare tumor with difficult diagnosis. This entity is highly aggressive with a poor prognosis. Clinical findings, histological analyses and the immunohistochemistry profile are required to achieve the correct diagnosis. Surgery was performed in most cases and the benefit of adjuvant therapy was not clear.
The authors declare no competing interests.
Amal Douida drafted the manuscript. All the authors have read and agreed to the final manuscript.
Table 1: some reported thyroid carcinoma with rhabdoid phenotype in the literature
Figure 1: heterogenous nodule with whitish and blackish areas
Figure 2: tumor composed of poorly differentiated large cells
Figure 3: rhabdoid cells with an abundant cytoplasm and eccentric nuclei containing distinct nucleoli
Figure 4: tumor infiltrated the perithyroid fat tissue
Figure 5: rhbadoid cells positive for CKAE1/AE3
Figure 6: rhabdoid cell showed immunoactivity for EMA
Figure 7: rhabdoid cells immunoreactive for Vimentin
- Beckwith JB, Palmer NF. Histopathology and prognosis of Wilms tumors: results from the First National Wilms´ Tumor Study. Cancer. 1978 May;41(5):1937-48. PubMed | Google Scholar
- Haas JE, Palmer NF, Weinberg AG, Beckwith JB. Ultrastructure of malignant rhabdoid tumor of the kidney, a distinctive renal tumor of children. Hum Pathol. 1981 Jul;12(7):646-57. PubMed | Google Scholar
- Biggs PJ, Garen PD, Powers JM, Garvin AJ. Malignant rhabdoid tumor of the central nervous system. Hum Pathol. 1987 Apr;18(4):332-7. PubMed | Google Scholar
- Lai ML, Faa G, Serra S, Senes G, Daniele GM, Boi F et al. Rhabdoid tumor of the thyroid gland: a variant of anaplastic carcinoma. Arch Pathol Lab Med. 2005 Mar;129(3):e55-7. PubMed | Google Scholar
- Sato K, Waseda R, Tatsuzawa Y, Soma R, Ueda Y, Katsuda S. Papillary thyroid carcinoma with anaplastic transformation showing a rhabdoid phenotype solely in the cervical lymph node metastasis. Pathol Res Pract. 2006;202(1):55-9. PubMed | Google Scholar
- Chetty R, Bhana B, Batitang S, Govender D. Lung carcinomas composed of rhabdoid cells. Eur J Surg Oncol. 1997 Oct;23(5):432-4. PubMed | Google Scholar
- Wick MR, Ritter JH, Dehner LP. Malignant rhabdoid tumors: a clinicopathologic review and conceptual discussion. Semin Diagn Pathol. 1995 Aug;12(3):233-48. PubMed | Google Scholar
- Borek BT, McKee PH, Freeman JA, Maguire B, Brander WL, Calonje E. Primary malignant melanoma with rhabdoid features: a histologic and immunocytochemical study of three cases. Am J Dermatopathol. 1998 Apr;20(2):123-7. PubMed | Google Scholar
- Chetty R. Rhabdoid tumors of the gastrointestinal tract. Am J Surg Pathol. 1994 Aug;18(8):858-9. PubMed | Google Scholar
- Parham DM, Peiper SC, Robicheaux G, Ribeiro RC, Douglass EC. Malignant rhabdoid tumor of the liver, evidence for epithelial differentiation. Arch Pathol Lab Med. 1988 Jan;112(1):61-4. PubMed | Google Scholar
- Chetty R, Govender D. Follicula thyroid carcinoma with rhabdoid phenotype. Virchows Arch. 1999 Aug;435(2):133-6. PubMed | Google Scholar
- Lu YT, Huang HI, Yang AH, Tai SK. Thyroid carcinoma with rhabdoid phenotype: Case report with review of the literature. Auris Nasus Larynx. 2016 Dec;43(6):706-9. PubMed | Google Scholar
- Carda C, Ferrer J, Vilanova M, Peydró A, Llombart-Bosch A. Anaplastic carcinoma of the thyroid with rhabdomyosarcomatous differentiation: a report of two cases. Virchows Arch. 2005 Jan;446(1):46-51. PubMed | Google Scholar
- Albores-Saavedra J, Sharma S. Poorly differentiated follicular thyroid carcinoma with rhabdoid phenotype: a clinicopathologic, immunohistochemical and electron microscopic study of two cases. Mod Pathol. 2001 Feb;14(2):98-104. PubMed | Google Scholar
- D´Antonio A, Orabona P, Caleo A, Addesso M, Liguori G, Boscaino A. Primary rhabdoid tumor of thyroid gland, description of a rare entity with molecular study. Pathol Int. 2010 Sep;60(9):644-6. PubMed | Google Scholar
- Agaimy A. The expanding family of SMARCB1(INI1)-deficient neoplasia: implications of phenotypic, biological and molecular heterogeneity. Adv Anat Pathol. 2014 Nov;21(6):394-410. PubMed | Google Scholar
- Sumida T, Hamakawa H, Imaoka M, Okamoto N, Takarada M, Tanioka H et al. A case of submandibular malignant rhabdoid tumor transformed from papillary thyroid carcinoma. Journal of Oral Pathology and Medicine. 2001;30(7):443-447. PubMed | Google Scholar
- Feng G, Laskin W B, Chou P M, Lin X. Anaplastic thyroid carcinoma with rhabdoid features. Diagn Cytopathol. 2015;43(5):416-420. PubMed | Google Scholar
- Assa SL. The Current Histologic Classification of Thyroid Cancer. Endocrinol Metab Clin North Am. 2019 Mar;48(1):1-22. PubMed | Google Scholar
- Pratima K, Shashikala K. Papillary carcinoma of thyroid with rhabdoid phenotype transformation: case report with review of the literature. International Journal of Science and Research (IJSR). 2018.
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures
Article metrics

