Soft tissue huge solitary neurofibroma in the sacral region without neurofibromatosis: a pediatric case report
Meriem Braiki, Mohamed Azzaza, Moncef Mokni, Khaled Sakly, Dorra Daly, Fethi Derbel
Corresponding author: Meriem Braiki, Department of Surgery, Sidi Bouzid Regional Hospital, Sidi Bouzid, Tunisia 
Received: 03 Mar 2020 - Accepted: 08 Mar 2020 - Published: 11 Mar 2020
Domain: Pediatric surgery
Keywords: Neurofibromas, soft tissue, surgery, histopathology
©Meriem Braiki et al. PAMJ Clinical Medicine (ISSN: 2707-2797). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Meriem Braiki et al. Soft tissue huge solitary neurofibroma in the sacral region without neurofibromatosis: a pediatric case report. PAMJ Clinical Medicine. 2020;2:96. [doi: 10.11604/pamj-cm.2020.2.96.22133]
Available online at: https://www.clinical-medicine.panafrican-med-journal.com//content/article/2/96/full
Case report 
Soft tissue huge solitary neurofibroma in the sacral region without neurofibromatosis: a pediatric case report
Soft tissue huge solitary neurofibroma in the sacral region without neurofibromatosis: a pediatric case report
Meriem Braiki1,&, Mohamed Azzaza2, Moncef Mokni3, Khaled Sakly3, Dorra Daly4, Fethi Derbel5
1Department of Surgery, Sidi Bouzid Regional Hospital, Sidi Bouzid, Tunisia, 2Department of Surgery, Sahloul University Hospital, Sousse, Tunisia, 3Department of Pathology, Farhat Hached Hospital, Sousse, Tunisia, 4Basic Center Health, Sidi Bouzid, Tunisia, 5Department of Surgery, Clinique Les Olivier, Sousse, Tunisia
&Corresponding author
Meriem Braiki, Department of Surgery, Sidi Bouzid Regional Hospital, Sidi Bouzid, Tunisia
Neurofibromas are thick and irregular benign neural sheath tumors touching the peripheral nerve and it may occur at any point along a nerve. Neurofibromas occur frequently as a neurofibromatosis manifestation and less commonly solitary, in unusual sites without neurofibromatosis. The imaging especially MRI is relatively helpful to determine radiological features necessary for the diagnosis, but the definitive diagnosis is established basing on the histopathological examination of the specimen. The primary therapeutic approach for neurofibromas is a complete surgical removal to prevent tumor recurrence. This article highlights a rare case of unusual soft tissue huge solitary neurofibroma in the sacral region without neurofibromatosis in a 12-old-girl.

Neurofibromas are the most common fibroblastic neoplasms affecting penipheral nerves [1]. These benign tumors are originating from different cells constituting the nerve sheath such as neurites, Schwann cells, and fibroblasts. However, the exact cell leading to neurofibroma occurrence has not been obviously identified [2]. Neurofibromas histologic findings vary from collagenous to myxoid matrix according to the neoplastic elements differentiation [1]. Typically, neurofibromas are described in patients with neurofibromatosis, known as, less commonly, neurofibromas may be reported as solitary lesions or as part of a generalized syndrome of neurofibromatosis. Rarely, multiple neurofibromas can occur without von Recklinghausen´s disease. Solitary neurofibroma is a localized tumor that generally occurs in patients without neurofibromatosis as the present case [3,4]. The reported case of soft tissue huge neurofibroma in the sacral region affecting an infant without neurofibromatosis is not well documented in the literature due to the rarity of such tumor at such site.

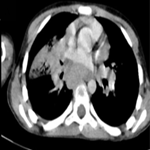
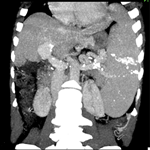
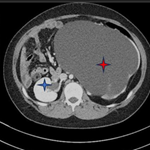
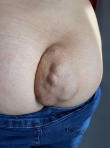
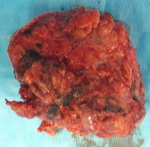
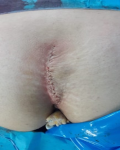
A 12-year-old girl was referred to the surgical department for low back painless swelling since the early childhood but neglected by the parents. The patient has unremarkable medical or surgical history. On physical examination, a huge swelling localized in the sacral region was clearly evident on inspection. A palpatory retrosacral mass was found, it was firm, nontender, well limited and measuring about 10*5cm (Figure 1). Neurological examination found no limb paresis. Routine laboratory tests were within normal limits. Magnetic resonance images (Figure 2) showed an 8*7*6cm retrosacral lobulated, non-encapsulated and lobulated mass. The lesion had intermediate signal intensity on T1-weighted images and bright intensity on T2-weighted images, with slight enhancement following the contrast administration. Intraoperatively, a well-circumscribed, encapsulated, firm and white-yellow mass was dissected and a complete excision of the mass was performed with macroscopic adjacent healthy tissue. The resected lesion was ovoid, weighed 300 grams and measured 9*7*6cm (Figure 3). Histopathological report confirmed the diagnosis of neurofibromas and revealed moderate cellularity with with elongated and fusiform cells containing several dark-stained nuclei embedded in a collagenous matrix. The post operative course was uneventful with good esthetic outcomes (Figure 4), and the patient was discharged on the postoperative 3th day with no neurological deficit. A follow-up period of 8 months has shown no evidence of tumor recurrence.
Neurofibromas account for 16-30% of all spinal tumors and 13.7% of spinal tumors originating from different elements of nerve sheath including Schwann cells, peri-neurial cells, and fibroblasts. Neurofibromas are most commonly localized in the thoracic region, followed by the cervical and lumbar sites. The sacral region is involved in only 1-5% of all spinal neurofibromas [2]. Neurofibromatosis type I, known as "von Recklinghausen disease", is a common genetic condition. An autosomal dominant pathology that affects the nervous system related to genetic disorder leading to a defective protein (neurofibromin) which is thought to act as a neoplasm suppressor. Several clinical features characterize the disease such as characterized pigmented skin lesions (café-au-lait macules), benign soft tissues tumors called neurofibromas, distinctive bone lesions and focal iris deformities. Neurofibromas are most commonly associated with neurofibromatosis type I [5]. Neurofibromas are frequently multiple and plexiform with a malignancy potential, they occur as a neurofibromatosis manifestation when they are deep and multiple. Malignant degeneration to neurofibrosarcoma occurs in 4-11% of patients with neurofibromatosis [1,6]. Solitary neurofibromas especially if cutaneous without neurofibromatosis are benign and rarely encountered [1]. The most common clinical presentation for neurofibromas is pain and radiculopathy due to compression of the involved nerve root [5]. Imaging is required for unusual tumor sites, for the tumor extent determination necessary to establish a proper surgical tumor management. On magnetic resonance imaging (MRI), neurofibromas may be inhomogeneous, tend to be isointense with muscle on T1-weighted images and show marked bright intensity with slight enhancement following the contrast administration on T2- weighted images [1,7]. Histologically, all neurofibromas are mixed tumors and are composed of non-neoplastic nerve elements including mast cells fibroblasts and sheath elements [8,9]. The diffrent cellular components of neurofibromas are embedded in an abundant collagenous and often myxoid extracellular matrix containing fibroblasts, Schwann cells, collagen and mucin [10]. The primary therapeutic option for neurofibromas is surgery. These tumors can be surgically removed and the surgical decision should be carefully made basing on the clinical and imaging features of the neoplasm. The complete surgical resection of the lesion is the treatment of choice for neurofibromas. A total lesion removal is necessary to prevent local recurrence and malignant transformation. The neurologic outcome is related to the tumor site and to weather the adjacent nerve root.
This article is dealing with a rare location of neurofibroma with particular features. Surgeons should be aware of this entity especially when the patient does not present neurofibromatosis manifestations. The exact diagnosis would be definitively determined by the histopathological examination of the specimen.
The authors declare no competing interests.
All the authors have read and agreed to the final manuscript.
Figure 1: clinical photo showing the lesion. Retrosacral huge mass measuring about 10x5cm
Figure 2: magnetic resonance images showed a 8x7x6 cm retrosacral lobulated, encapsulated and well-limited mass. The lesion shows slight enhancement following the contrast administration
Figure 3: the resected lesion was ovoid, weighed 300 grams and measured 9x7x6cm
Figure 4: post operative appearance showing esthetic result
- Barboriak DP, Rivitz SM, Chew FS. Sacral neurofibroma. AJR Am J Roentgenol. 1992 Sep;159(3):600. PubMed | Google Scholar
- Topsakal C, Erol FS, Ozercan I, Murat A, Gurates B. Presacral solitary giant neurofibroma without neurofibromatosis type 1 presenting as pelvic mass-case report. Neurol Med Chir (Tokyo). 2001;41(12):620-5. PubMed | Google Scholar
- Anjali N, Susmita S, Vanita R, Puja B. Intraoral solitary neurofi broma in an infant. Journal of Oral and Maxillo Facial Pathology. 2008;12(2):75-78. Google Scholar
- Sivapathasundaram B, Lavanya S, Deeplakshmi, Saravanakumar R, Ahathya RS. Solitary Neurofibroma of the gingival. J Oral Maxillofac Pathol. 2004;8(2):107-9. Google Scholar
- Brian CF, Nathan S. Neurofibroma of the anterior leg: a case report. The Foot and Ankle Online Journal. 2012;5(6). Google Scholar
- Meek RM, Sharma H, Jane MJ, Raby N, Macduff E, Reid R. Solitary intraosseous schwannoma of the metatarsal bone: case report. Foot Ankle Int. 2007;28(7):845-8. PubMed | Google Scholar
- Breidahl WH, Khangure MS. MRI of lumbar and sacral plexus nerve sheath tumours. Australas Radiol. 1991;35(2):140-4. PubMed | Google Scholar
- Carroll SL, Ratner N. How does the Schwann cell lineage form tumors in NF1. Glia. 2008;56(14):1590-605. PubMed | Google Scholar
- Le LQ, Shipman T, Burns DK, Parada LF. Cell of origin and microenvironment contribution for NF1-associated dermal neurofibromas. Cell Stem Cell. 2009;4(5):453-63. PubMed | Google Scholar
- Ortonne N, Wolkenstein P, Blakeley JO, Korf B, Plotkin SR, Riccardi VM et al. Cutaneous neurofibromas: current clinical and pathologic issues. Neurology. 2018 Jul 10;91(2 Suppl 1):S5-S13. PubMed | Google Scholar
Search

This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures

Article metrics
Recently from the PAMJ-CM