
Idiopathic pleuroparenchymal fibroelastosis: a rare case report
Hajar Hamri, Zakaria Toufga, Sanae Chaoui, Hounaida Jerguigue, Rachida Latib, Youssef Omor
Corresponding author: Hajar Hamri, Imaging Department, National Institute of Oncology. Mohammed V University, Rabat, Morocco 
Received: 29 Apr 2020 - Accepted: 18 May 2020 - Published: 01 Jun 2020
Domain: Radiology,Pulmonology
Keywords: Fibroelastosis, pleura, lung, interstitial pneumonia
©Hajar Hamri et al. PAMJ Clinical Medicine (ISSN: 2707-2797). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Hajar Hamri et al. Idiopathic pleuroparenchymal fibroelastosis: a rare case report. PAMJ Clinical Medicine. 2020;3:34. [doi: 10.11604/pamj-cm.2020.3.34.23176]
Available online at: https://www.clinical-medicine.panafrican-med-journal.com//content/article/3/34/full
Case report 

Idiopathic pleuroparenchymal fibroelastosis: a rare case report
Idiopathic pleuroparenchymal fibroelastosis: a rare case report
Hajar Hamri1,&, Zakaria Toufga1, Sanae Chaoui1, Hounaida Jerguigue1, Rachida Latib1, Youssef Omor1
1Imaging Department, National Institute of Oncology, Mohammed V University, Rabat, Morocco
&Corresponding author
Hajar Hamri, Imaging Department, National Institute of Oncology. Mohammed V University, Rabat, Morocco
Idiopathic pleuroparenchymal fibroelastosis (IPPFE) is a rare clinical-pathologic entity characterized by fibrotic thickening of the pleural and subpleural parenchyma predominantly in the upper lobes. The clinical course of the disease is progressive and the prognosis is poor. The only therapeutic option is lung transplantation. We report a case fulfilling the criteria of IPPFE in a 47-year-old female, in order to review the CT and clinical aspects as well as the differential diagnoses of this disorder.

Idiopathic pleuroparenchymal fibroelastosis (IPPFE) is a rare clinical-pathological entity, distinguished from other types of idiopathic interstitial pneumonia [1,2]. IPPFE is characterized by fibrotic thickening of the pleural and sub-pleural parenchyma mainly of the upper lobes. Generally, the diagnosis is suspected in imaging and confirmed by histology. We report the case of a 47-year-old female whose clinical and radiological findings were in favor of IPPFE that has been proven histologically.

A 47-year-old female non-smoker and with no particular medical history, presenting for approximately 2 years, progressive dyspnea and a dry cough with the notion of weight loss estimated at 10kg. Infectious laboratory tests, including tuberculosis, were negative and a restrictive lung disease was identified in pulmonary function tests. The chest CT-scan showed bilateral pleural thickening with subpleural condensations of the apex and loss of volume in both upper lobes (Figure 1, Figure 2) which were suggestive of pleuroparenchymal fibroelastosis. The lung biopsy performed following the CT scan results with anatomopathological analysis using a specific coloring of elastin fibers showed a fibrosis of the visceral pleura with subpleural intra-alveolar fibrosis and lymphoplasmocytic infiltrates, confirming the diagnosis.
Pleuroparenchymal fibroelastosis is a rare entity identified and classified as idiopathic interstitial lung disease according to the American Thoracic Society/European Respiratory Society Statement published in June 2013 [1]. It affects both sexes equally with a median age of 57 ans years [2]. A history of pneumothorax and recurrent infections are common, a minority has familial interstitial lung disease and nonspecific autoantibodies but most cases are idiopathic [1]. Clinically, dyspnea is constant and constitutes the main reason for consultation, cough is frequent (> 50%), weight loss is almost always present, as well as chest pain. A small anteroposterior chest diameter that appears gradually during the disease is quite characteristic. Pneumothorax is very common and sometimes bilateral [3]. The histological criteria of typical IPPFE as defined by Kusagaya et al. [4] include intense fibrosis of the visceral pleura; prominent, homogenous, subpleural fibroelastosis; sparing of the parenchyma distant from the pleura; mild, patchy lymphoplasmocytic infiltrates; and presence of small numbers of fibroblastic foci. Von der Thüsen [5] has defined additional histological criteria to establish a diagnosis of a pattern of fibrosis which is “consistent with” IPPFE, being intra-alveolar fibrosis as above, but not associated with significant pleural fibrosis, or not predominantly beneath the pleura, or not in upper lobe biopsy.
In high-resolution CT imaging, IPPFE is characterized by dense pleural and subpleural consolidation which may be responsible for traction bronchiectasis, architectural distortion, and upper lobe volume loss [1,6]. Pleural thickening can be observed along the fissures. The lesions can be modest initially. The absence of pleural involvement of the bases is characteristic, but as the disease progresses, the opacities may extend to the adjacent lobes [6]. The radiological criteria [5] proposed are: a definite diagnosis when there is a pleural thickening and a subpleural fibrosis centered in the upper lobes and consistent with the diagnosis when there is u upper lobe pleural thickening and subpleural fibrosis, but distribution of changes not concentrated in upper lobes, or presence of features of coexistent disease elsewhere. The differential radiological diagnosis of IPPFE includes: asbestosis, connective tissue diseases, advanced fibrosing sarcoidosis, radiation or drug-induced lung disease, chronic hypersensitivity pneumonitis, and apical caps. In hypersensitivity pneumonitis (HP) lesions are bronchiolocentric and bronchiolization of the centrilobular airways is prominent [6]. Apical caps may be difficult to distinguish from IPPFE and probably the difference is mainly quantitative. The differential diagnosis could be often reached on the basis of clinical and radiological features: IPPFE is an interstitial lung disease in opposite to apical cap [7,8]. The evolution of this affection is progressive in 60% of patients and the prognosis is poor with death due to the disease in 40% [9,10]. Given the low efficacy of immunosuppressive treatments in these cases, the only therapeutic option remains lung transplantation [6].
Idiopathic PPFE is a highly progressive disease, of quite specific radiological and histological presentation, which is distinguished from other types of interstitial lung disease. This diagnosis should be suspected when radiological evidence is not compatible with well-defined idiopathic pneumonias affecting the upper lobes.
The authors declare no competing interests.
All authors have contributed to the work and write-up of the manuscript. All the authors have read and agreed to the final manuscript.
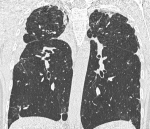
Figure 1: pleuroparenchymal fibroelastosis. High-resolution CT scan: coronal image shows pleural and subpleural thickening with moderate fibrotic changes in the marginal parenchyma clearly predominant in the upper lobes with architectural distortion and volume loss in the upper lobes
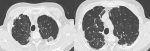
Figure 2: pleuroparenchymal fibroelastosis. High-resolution CT scan: axial images through the upper lobes shows a pleural thickening and subpleural consolidation with traction bronchiectasis
- Travis WD, Costabel U, Hansell DM, King TE Jr, Lynch DA, Nicholson AG et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013 Sep 15;188(6):733-48. PubMed | Google Scholar
- Kadoch MA, Cham MD, Beasley MB, Ward TJ, Jacobi AH, Eber CD et al. Idiopathic interstitial pneumonias: a radiology-pathology correlation based on the revised 2013 American Thoracic Society European Respiratory Society classification system. Current Problems in Diagnostic Radiology. 2015 Jan-Feb;44(1):15-25. PubMed | Google Scholar
- Goyard C, Bonniaud P. Fibroélastose pleuro-parenchymateuse. Revue des Maladies Respiratoires Actualités. 2016;8:108-110. Google Scholar
- Hideki Kusagaya, Yutaro Nakamura, Masato Kono, Yusuke Kaida, Shigeki Kuroishi, Noriyuki Enomoto et al. Idiopathic pleuroparenchymal fibroelastosis: consideration of a clinicopathological entity in a series of Japanese patients. BMC Pulm Med. 2012;12:72. PubMed | Google Scholar
- Von der Thüsen JH. Pleuroparenchymal fibroelastosis: its pathological characteristics. Curr Respir Med Rev. 2013 Aug;9(4):238-247. PubMed | Google Scholar
- Piciucchi S, Tomassetti S, Casoni G, Sverzellati N, Carloni A, Dubini A et al. High resolution CT and histological findings in idiopathic Pleuroparenchymal fibroelastosis: features and differential diagnosis. Respir Res. 2011;12(1):111. PubMed | Google Scholar
- Becker CD, Gil J, Padilla M. Idiopathic pleuroparenchymal fibroelastosis: an unrecognized or misdiagnosed entity. Mod Pathol. 2008 Jun;21(6):784-7. PubMed | Google Scholar
- Rozin GF, Gomes MM, Parra ER, Kairalla RA, de Carvalho CR, Capelozzi VL. Collagen and elastic system in the remodelling process of major types of idiopathic interstitial pneumonias (IIP). Histopathology. 2005 Apr;46(4):413-21. PubMed | Google Scholar
- Kentaro Watanabe. Pleuroparenchymal fibroelastosis: its clinical characteristics. Curr Respir Med Rev. 2013;9:229-237. Google Scholar
- Reddy TL, Tominaga M, Hansell DM, von der Thusen J, Rassl D, Parfrey H et al. Pleuroparenchymal fibroelastosis: a spectrum of histopathological and imaging phenotypes. Eur Respir J. 2012 Aug;40(2):377-85. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures
Article metrics

