
Le pronostic d´un lymphome anaplasique à grandes cellules chez une jeune fille de 15 ans aggravé par un syndrome d´activation marophagique. a propos d´un cas et revue de la littérature
Jihane Toughza, Aomar Agadr, Mohamed Miguil, Youssef Tijani
Corresponding author: Jihane Toughza, Département d´Oncologie et Hématologie Pédiatrique, Hôpital International Universitaire Cheikh Khalifa, Faculté de Médecine, Université Mohammed VI des Sciences de la Santé, Casablanca, Maroc 
Received: 15 Jun 2020 - Accepted: 24 Jul 2020 - Published: 25 Sep 2020
Domain: Médecine de s’intensifs, hématologie pédiatrique, chirurgie vasculaire
Keywords: Lymphome anaplasique, adolescente, syndrome activation macrophagique
©Jihane Toughza et al. PAMJ Clinical Medicine (ISSN: 2707-2797). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Jihane Toughza et al. Le pronostic d´un lymphome anaplasique à grandes cellules chez une jeune fille de 15 ans aggravé par un syndrome d´activation marophagique. a propos d´un cas et revue de la littérature. PAMJ Clinical Medicine. 2020;4:40. [doi: 10.11604/pamj-cm.2020.4.40.24334]
Available online at: https://www.clinical-medicine.panafrican-med-journal.com//content/article/4/40/full
Case report 

Le pronostic d´un lymphome anaplasique à grandes cellules chez une jeune fille de 15 ans aggravé par un syndrome d´activation marophagique. a propos d´un cas et revue de la littérature
Le pronostic d´un lymphome anaplasique à grandes cellules chez une jeune fille de 15 ans aggravé par un syndrome d´activation marophagique : à propos d´un cas et revue de la littérature
Prognosis of a 15-year old girl with anaplastic large cell lymphoma worsened by macrophage activation syndrome: case study and literature review
Jihane Toughza1,&, Aomar Agadr2, Mohamed Miguil3, Youssef Tijani4
&Auteur correspondant
Une jeune fille de 15 ans, se présente aux urgences pédiatriques pour des douleurs abdominales. Il s´avère à l´interrogatoire que la symptomatologie remonte à 1 mois. La patiente a bénéficié d´une appendicectomie 15 jours auparavant n´ayant pas amélioré les douleurs abdominales. La patiente présente un lymphome anaplasique de haut grade accompagné d´un syndrome d´activation macrophagique. Le bilan radiologique révèle des adénopathies dont la plus importante en iliaque droite. La biopsie sous échographie confirme la nature tumorale. Seule analyse immuno-histochimique permet d´établir le diagnostic de lymphome anaplasique de haut grade. L´association de ces deux entités est peu décrite et source d´errance ou de retard du diagnostic pour une pathologie engagent le pronostic vital.
The study involved a 15-year old girl presenting to the Pediatric Emergency Department with abdominal pain. During the initial interview, the patient reported first symptoms one month before. The patient had undergone appendectomy 15 days earlier, without improvement in abdominal pain. The patient presented with high grade anaplastic lymphoma associated with macrophage activation syndrome. Radiological evaluation revealed lymphadenopathies, mostly in the right iliac lymph nodes. Ultrasound-guided biopsy confirmed the diagnosis of tumor. Only immunohistochemical analysis established the diagnosis of high grade anaplastic lymphoma. The association between these two entities has been little described in the literature, leading to mistakes in diagnosis or delay in diagnosis in patients with this life-threatening disease.
Key words: Anaplastic lymphoma, adolescent, macrophage activation syndrome

Le lymphome anaplasique à grandes cellules (LAGC) est un lymphome non-Hodgkinien, à cellules T périphériques. Cette entité de lymphome est rare agressive et affecte les ganglions lymphatiques et les sites extranodaux [1,2]. Son mode de présentation clinique peut simuler d´autres pathologies prêtant alors à confusion [3]. Le diagnostic est histologique, avec les techniques immuno-histochimiques et celles de la biologie moléculaire. L´association de ce type de lymphome au syndrome d´activation macrophagique majore le risque d´errance de diagnostic, mettant en jeu le pronostic vital du patient. Dans ce travail, nous rapportons l'observation d'un cas de LAGC chez une jeune fille de 15 ans, dont l´intérêt porte sur la discussion diagnostique qu'elle a soulevé et sur la prise en charge thérapeutique. Nous rappelons les difficultés posées par le diagnostic de LAGC associé à un syndrome d´activation macrophagique à cet âge.

Une jeune fille âgée de 15 ans, sans antécédents personnel ou familial notable, est admise le 16 février 2017 pour des douleurs abdominales évoluant depuis 1 mois. Dans ses antécédents, on note une appendicectomie par cœlioscopie 15 jours avant l´hospitalisation, sans signe de malignité à l´examen anatomopathologique, et n´ayant pas amélioré les douleurs abdominales. Un scanner abdominopelvien a été réalisé avant l´hospitalisation montrant des adénopathies lomboaortiques et iliaques. Le tableau clinique à l´arrivée est dominé par des douleurs abdominales d´abord généralisées, puis secondairement localisées au niveau splénique. Sur le plan biologique, un Quantiferon et une IDR sont revenus négatifs, et ont permis de récuser le diagnostic de tuberculose péritonéale. La NFS initiale était en faveur d´une thrombopénie à 69 000/mm3 associée à une hyperleucocytose à 22 000/mm3 à prédominance à polynucléaires neutrophile à 18 460/mm3.
La CRP était à 370 mg/l. La pièce d´appendicectomie a été relue, sans signe de malignité retrouvé, et une biopsie sous échographie de l´adénopathie iliaque externe droite a été réalisée. L´examen anatomo-pathologique de cette biopsie sous échographie a proposé morphologiquement deux diagnostics différentiels. En effet, il s´agirait d´une prolifération tumorale indifférenciée dont la nature lymphomateuse ou carcinomateuse ne pouvait être précisée. L´examen par Fish sur bloc en paraffine a mis en évidence une absence de réarrangement Bcl-2, une absence de réarrangement d´IgH/Bcl-6, et une absence de remaniement et/ou d´amplification du locus c-MYC/IgH. Le myélogramme au niveau des deux épines iliaques postérieures est revenu normal. Elle a présenté ensuite un tableau fugace de douleurs paroxystiques du membre inférieur droit avec des troubles de la sensibilité, des troubles de la conscience avec un AVC massif ischémique puis des signes d´ischémie du membre inférieur droit.
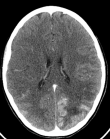
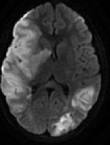
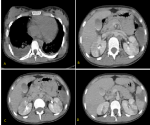
A l´examen clinique, la patiente était fébrile, une splénomégalie, une adénopathie sus claviculaire gauche significative ont été retrouvées. Elle a été transférée en urgence en réanimation. La NFS retrouvait une anémie à 9.1 g/dl, une thrombopénie à 90 000/mm3, une hyperleucocytose à 34240/mm3 à prédominance polynucléaires neutrophiles à 29070/mm3, le bilan hépatite révèle une cytolyse (ASAT à 110UI/l) et les LDH sont à 1091 UI/l, évoquant un syndrome d´activation macrophagique responsable des lésions vasculaires. Une ponction lombaire est revenue négative, et la tomodensitométrie TDM cérébrale (Figure 1) évoquait une encéphalite infectieuse. L´IRM cérébrale (Figure 2) était en faveur de multiples lésions ischémiques cérébrales bilatérales dont la plus volumineuse est temporo-fronto-pariétale droite secondaire à une occlusion de l´artère sylvienne homolatérale. Étant sous antibiotiques pour hyperthermie, l´aciclovir a été rajouté. Un TDM cervico-thoraco-abdominal (Figure 3) a mis en évidence des polyadénopathies sus et sous diaphragmatiques. Une pleuropneumopathie bilatérale prédominant à droite. Des plages hypodenses spléniques et rénales pouvant être en rapport avec des infarctus. Une thrombose vasculaire en regard de la bifurcation aortique et l´artère iliaque primitive droite.
Devant une ischémie du membre inférieur droit, la patiente a bénéficié en urgence d´une chirurgie de revascularisation avec le prélèvement diagnostic d´une adénopathie. Une corticothérapie à haute dose associée à une héparinothérapie efficace a été administrée pour la prise en charge du syndrome d´activation macrophagique juste après le prélèvement de l´adénopathie. Devant l´évolution très rapide du tableau clinique, la patiente a reçu une cure de chimiothérapie à base d´Endoxan en urgence associée à une corticothérapie à haute dose en attente des résultats définitifs de l´immunohistochimie. La patiente est décédée dans les suites immédiates de la première cure de chimiothérapie, dans un tableau de probable syndrome d´activation macrophagique avec défaillance multiviscérale. L´étude immunohistochimique a mise en évidence des cellules tumorales exprimant le CD30 et ALK1. Le CD20 et le CD3 marquent le fond lymphocytaire. Le CD15 est négatif. Le CD68 marque les histiocytes. L´anti-LMP1 est négatif. Il s´agit donc d´un lymphome anaplasique à grandes cellules ALK+, dont le diagnostic final était en post-mortem.
L´intérêt de ce cas réside dans la rareté du diagnostic, le mode de présentation et d´évolution qui prêtait à confusion entre lymphome intravasculaire et lymphome anaplasique. Le lymphome anaplasique à grandes cellules (LAGC) est un lymphome non-Hodgkinien à cellules T périphériques, rare et agressif, appartenant au groupe des syndromes lymphoprolifératifs CD30 positifs et affectant les ganglions lymphatiques et les sites extranodaux [1,2]. Le LAGC compte pour environ 3% des lymphomes non-hodgkiniens de l'adulte et pour 10 à 20% des lymphomes de l'enfant [3]. Les circonstances de découvertes sont variables avec habituellement un tableau de polyadénopathies et d´altération de l´état général. Ils sont rarement décrits en association avec un syndrome d´activation macrophagique. Sa prévalence est inconnue. Il comprend deux sous-types, selon l'expression d'une protéine appelée kinase du lymphome anaplasique (ALK) [4]: le LAGC ALK positif et le LAGC ALK négatif. Le sous-type ALK positif affecte généralement les enfants et les jeunes adultes. Le sous-type ALK négatif est retrouvé plus souvent chez les patients plus âgés, à partir de l'âge de 40 ans [5]. L'ALCL systémique a une distribution bimodale, avec un pic dans l'enfance et un autre à la fin de l'âge adulte. Le LAGC est caractérisé par l'atteinte des ganglions lymphatiques périphériques, médiastinaux et abdominaux. Il se manifeste par le développement d'adénopathies non douloureuses, surtout au niveau du cou ou des aisselles (ganglions lymphatiques axillaires). Les symptômes généraux incluent une anorexie et une fatigue, ainsi qu'une fièvre, une perte de poids et des sueurs nocturnes (symptômes B). L'atteinte médiastinale se manifeste par une toux, une dyspnée et/ou un œdème.
Le LAGC peut aussi s'étendre à des sites extranodaux tels que les os, la moelle osseuse, le tissu sous-cutané, les poumons, la rate et le foie. L'étiologie de la maladie est inconnue. Dans le sous-type ALK positif, le gène ALK (2p23) du récepteur de la tyrosine kinase du lymphome anaplasique est surexprimé, dû à une translocation t (2;5)(p23;q35). Le diagnostic est basé sur l'examen physique, les antécédents médicaux et est confirmé par l'examen histopathologique et immunohistochimique d'une biopsie de ganglion lymphatique. La biopsie montre une tumeur cohésive, généralement avec une atteinte des sinus et une cytologie anaplasique (grandes cellules atypiques avec un cytoplasme abondant, des nucléoles proéminents et un noyau en forme de rein ou en forme de fer à cheval), l'expression membranaire constante de l'antigène CD30, l'expression de l'antigène épithélial membranaire (EMA) dans la majorité des cas, et l'expression de CD3, CD5 ou CD2 (surtout en cas de LAGC à ALK négatif). La protéine ALK est détectée par immunohistochimie pour le sous-type ALK positif. Des tests additionnels incluent des analyses de sang et de moelle osseuse, ainsi qu'une imagerie (rayons X, tomodensitométrie, PET scan, et IRM) pour la maladie osseuse. Le traitement pédiatrique comporte une corticothérapie associée à une polychimiothérapie basée sur l'anthracycline, le cyclophosphamide, le méthotrexate, l'étoposide, l´holoxan et la cytarabine. La chimiothérapie à haute dose suivie par une transplantation autologue de cellules souches peut aussi être réalisée, généralement en cas de récidive ou en première ligne de traitement en cas de pronostic défavorable. La thérapie conjuguée anticorps-médicament (brentuximab velotin) peut être prescrite lorsqu'au moins un régime de chimiothérapie a échoué.
Avec le traitement, les patients ALK positifs (70 à 80% de survie à 5 ans) ont un meilleur pronostic que les patients ALK négatifs (33 à 49% de survie à 5 ans). La récidive appauvrit le prognostic [6]. Le pronostic peut être cependant péjoratif même chez les patients ALK+(6). Des sous-groupes morphologiques avec un composant à petite cellule (SC) ou lymphohistiocytique seraient de moins bon pronostic [6]. Le syndrome hémophagocytaire est responsable d´une activation des macrophages qui ingèrent les éléments figurés du sang (globules rouges, leucocytes, plaquettes). Dans sa forme habituelle, le syndrome comprend une profonde altération de l´état général, une hépatite, une pancytopénie, une hypofibrinémie et une hyperferritinémie importante. Il apparaît comme une voie finale commune de déficits immunitaires primitifs, de syndromes lymphoprolifératifs, de certaines infections, voire de réactions médicamenteuses ou de maladies systémiques. Henter et al. [7] distinguent les formes primitives et héréditaires de syndrome hémophagocytaire, représentées par la lymphohistiocytose hémophagocytaire familiale ou sporadique (regroupées sous le terme général de lymphohistiocytose hémophagocytaire) survenant dans l´enfance, et les formes secondaires habituellement associées à des thérapeutiques immunosuppressives, une affection maligne, auto-immune ou infectieuse [7].
Les hémopathies malignes représentent l´une des étiologies majeures du syndrome hémophagocytaire. Parmi celles-ci, les lymphomes non-Hodgkiniens, notamment de phénotype T anaplasique CD30, prédominent largement. L´association entre certains lymphomes et un syndrome d´activation macrophagique est classique chez l´adulte mais reste beaucoup plus rarement décrite dans le cadre des lymphomes anaplasiques à grandes cellules chez l´enfant [8,9]. Les difficultés diagnostiques pour le pathologiste sont peu abordées dans la littérature. Nous avons répertorié dans la littérature 10 cas distincts de lymphome anaplasique à grandes cellules associés à un syndrome d´activation macrophagique révélateur du diagnostic (Tableau 1) [8,10-16] . Il s´agit de lymphomes pédiatriques dans 8 cas. Dans la majorité des cas, le diagnostic d´une pathologie infectieuse était porté en premier lieu. Devant l´aggravation des symptômes sous traitement anti-infectieux et l´apparition d´adénopathies, le diagnostic était rétabli secondairement par l´examen histologique ganglionnaire. Florena et al. [17], dans son article suggère qu´un syndrome hémophagocytaire masque très souvent une pathologie maligne sous-jacente. La mise en jeu du pronostic vital impose un diagnostic précoce, la recherche systématique d´une cause et la mise en route d´un traitement idéalement étiologique [18].
LAGC est une entité rare à présentation clinique parfois aspécifique, simulant d´autres pathologies de caractère et de pronostic différents, prêtant alors à confusion. Le diagnostic est histologique et se base sur les techniques immuno-histochimiques notamment avec les anticorps anti-CD30 et anti-ALK, et plus récemment sur celles de la biologie moléculaire. Le traitement n´est pas encore codifié mais le pronostic peut être dramatique. L´association entre un syndrome d´activation macrophagique et un lymphome anaplasique à grandes cellules est une source d´errance diagnostique.
Les auteurs déclarent ne pas avoir de conflits d´intérêts en relation avec cet article.
Tous les auteurs ont contribués à la rédaction de l´article.
Tableau 1: les cas précédemment publiés associant syndrome d´activation macrophagique (HPS) à un lymphome anaplasique à grandes cellules (ALCL)
Figure 1: TDM cérébrale
Figure 2: IRM cérébrale
Figure 3: TDM TAP
- Wright D, McKeever P, Carter R. Childhood non-Hodgkin lymphomas in the United Kingdom: findings from the UK Children's Cancer Study Group.. J Clin Pathol. 1997 Feb;50(2):128-34. PubMed | Google Scholar
- Lim MS, de Leval L, Quintanilla-Martinez L. Commentary on the 2008 WHO classification of mature T- and NK-cell neoplasms. J Hematop. 2009 Jun 27;2(2):65-73. PubMed | Google Scholar
- Nagai K, Suyama Y, Koga D, Nishi M, Iida C, Tashiro K et al. Anaplastic lymphoma kinase-positive anaplastic large cell lymphoma with cardiac metastasis and arterial tumor embolisms during first-course chemotherapy. Case Rep Oncol. 2016 Aug 17;9(2):440-6. PubMed | Google Scholar
- Cluzeau T, Pécuchet N, Mounier N, Vignot S. Implications d´ALK (anaplastic lymphoma kinase) en oncohématologie. Bull Cancer (Paris). 2010;97(8):991-996. PubMed | Google Scholar
- Pomari E, Basso G, Bresolin S, Pillon M, Carraro E, d´Amore ES et al. NPM-ALK expression levels identify two distinct subtypes of paediatric anaplastic large cell lymphoma. Leukemia. 2017 Feb;31(2):498-501. PubMed | Google Scholar
- Lamant L, McCarthy K, d´Amore E, Klapper W, Nakagawa A, Fraga M et al. Prognostic impact of morphologic and phenotypic features of childhood ALK-positive anaplastic large-cell lymphoma: results of the ALCL99 study. J Clin Oncol Off J Am Soc Clin Oncol. 2011 Dec 10;29(35):4669-7. PubMed | Google Scholar
- Henter J-I, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007 Feb 1;48(2):124-31. PubMed | Google Scholar
- Blatt J, Weston B, Belhorn T, Hamrick H, Maia D. Childhood non-Hodgkin lymphoma presenting as hemophagocytic syndrome. Pediatric Hematology and Oncology. 2002;19(1):45-9. PubMed | Google Scholar
- Sizaret D, Lecointre C, Kerdraon R, Bléchet C, Bonneau C, Alexis Met al. Hemophagocytic lymphohistiocytosis associated with a lymphohistiocytic pattern anaplastic large cell lymphoma: a case report. annales de pathologie. 2013 Aug;33(4):278-82. PubMed | Google Scholar
- Cho SG, Koh YB, Chang HS, Park G, Kang CS, Park JW et al. Successful treatment with splenectomy and interferon alpha against recurred hemophagocytic syndrome in remission state of anaplastic large cell lymphoma following high-dose therapy and autologous peripheral blood stem cell transplantation. European Journal of Haematology. 2005 Mar;74(3):259-62. PubMed | Google Scholar
- Krenova Z, Sterba J, Blatny J, Kren L, Slany J. A case of anaplastic large cell lymphoma-induced hemophagocytic lymphohistiocytosis in an adolescent female. Pediatr Blood Cancer. 2007 Dec;49(7):1056. PubMed | Google Scholar
- Sevilla DW, Choi JK, Gong JZ. Mediastinal adenopathy, lung infiltrates, and hemophagocytosis: unusual manifestation of pediatric anaplastic large cell lymphoma: report of two cases. Am J Clin Pathol. 2007 Mar;127(3):458-64. PubMed | Google Scholar
- Shimada A, Kato M, Tamura K, Hirato J, Kanegane H, Takechi Y et al. Hemophagocytic Lymphohistiocytosis Associated With Uncontrolled Inflammatory Cytokinemia and Chemokinemia was caused by systemic anaplastic large cell lymphoma: a case report and review of the literature. J Pediatr Hematol Oncol. 2008 Oct;30(10):785-7. PubMed | Google Scholar
- Sovinz P, Lackner H, Schwinger W, Benesch M, Urban C, Beham-Schmid C. Anaplastic large cell lymphoma presenting as hemophagocytic syndrome in an adolescent. Pediatr Blood Cancer. 2007 Dec;49(7):1057. PubMed | Google Scholar
- Khor TS, Alessandri AJ, Jevon GP. Infant anaplastic large cell lymphoma with hemophagocytic syndrome. Pediatr Dev Pathol Off J Soc Pediatr Pathol Paediatr Pathol Soc. 2010 Feb;13(1):72-6. PubMed | Google Scholar
- Peluso MJ, Chia D, Sheen W, Hutchinson C, Barakat L. Infectious Infectious mimicry complicates diagnosis in hemophagocytic syndrome caused by anaplastic large cell lymphoma. Case Rep Med. Case Rep Med. 2012;2012:968706. PubMed | Google Scholar
- Florena AM, Lanitto E, Quintini G, Franco V. Bone marrow biopsy in hemophagocytic syndrome. Virchows Arch. 2002 Oct;441(4):335-44. PubMed | Google Scholar
- Balwierz W, Czogala M, Czepko E. Anaplastic large cell lymphoma associated with hemophagocytic lymphohistiocytosis: a case report and review of the literature. Przegl Lek. 2010;67(6):436-8. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures
Article metrics

