
Desmoid tumor of the abdominal wall: case report
Kaoutar Achehboune, Hanane Baybay, Khadija Issoual, Sara Elloudi, Fatima Zahra Mernissi
Corresponding author: Achehboune Kaoutar, Department of Dermatology, University Teaching Hospital, Hassan II, Fez , Morocco 
Received: 06 Mar 2020 - Accepted: 02 Nov 2020 - Published: 03 Nov 2020
Domain: Dermatology,Oncology,General surgery
Keywords: Desmoid, fibrous tumor, abdominal wall
©Kaoutar Achehboune et al. PAMJ Clinical Medicine (ISSN: 2707-2797). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Kaoutar Achehboune et al. Desmoid tumor of the abdominal wall: case report. PAMJ Clinical Medicine. 2020;4:78. [doi: 10.11604/pamj-cm.2020.4.78.22201]
Available online at: https://www.clinical-medicine.panafrican-med-journal.com//content/article/4/78/full
Case report 

Desmoid tumor of the abdominal wall: case report
Desmoid tumor of the abdominal wall: case report
Kaoutar Achehboune1,&, Hanane Baybay1, Khadija Issoual1, Sara Elloudi, Fatima Zahra Mernissi1
&Corresponding author
Desmoid tumors are rare fibrous tumors of the soft tissues. Develops on the abdominal wall, in the abdomen, or extra-abdominal. Parietal location is very common among women. Hormonal factors are incriminated. They are rare lesions without any metastatic potential a strong tendency to invade locally and to recur. Hormonal factors are incriminated in this pathology. Imaging can characterize these tumors and their reports and guide the biopsy that is the key to diagnosis. The treatment is based on first-line surgery, but in cases where excision margins are reached, local recurrence or unresectable tumors, other therapeutic alterations are needed such as radiotherapy and chemotherapy and others. We report a periumbilical case in perimenopause.

Desmoides tumors are rare fibrous tumors of the soft tissues. They lie in the abdominal wall, in the abdomen, or extra-abdominal. Parietal location is very common among women. Hormonal factors are thus incriminated. We report a peri-umbilical case.

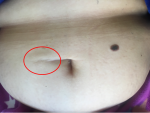
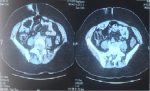
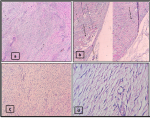
A 45 years old woman. On oral contraception, had no remarkable medical, traumatic or familial history, consulted for a nodular lesion in the peri-umbilical region (Figure 1). The nodule is poorly limited by 3 cm fixed in the deep plane and mobile at the superficial level without opposing inflammatory signs, slightly painful on palpation, without palpable intra-abdominal mass. The abdominopelvic scanner had revealed a suspicious parietal tumor (Figure 2). A biopsy revealed that an epidermis is of normal thickness, slightly papillomatous and surmounted by an orthokeratotic keratosis (Figure 3). The papillary dermis houses some congestive capillaries. The reticular dermis is fibrous and punctuated by some discrete mononuclear inflammatory infiltrates. This fibrosis extends to the hypodermis with mesenchymal tumor proliferation of moderate and heterogeneous density. It is made of fusiform cells of fibroblastic or myofibroblastic shape with elongated nuclei sometimes ovoid atypical and provided with a small nucleolus. These cells are arranged in long divergent bundles interwoven with corrugated collagen fibers. The vessels of medium abundance are sometimes surrounded by a light halot and a minimal lymphocytic infiltrate. Immunohistochemistry (Figure 4); the tumor expressed anti-smooth muscle actin antibody, anti Beta catenin antibody with nuclear labeling, Anti PS100 antibody (Polyclonal, DAKO) was not expressed by tumor proliferation and thus the morphological appearance and immunohistochemical data are suggestive desmoid fibromatosis. Surgical excision was performed with wide margins. The evolution was good without recurrence after 12 months of follow-up.
Desmoid tumors are rare fibrous soft tissue tumors developed from connective tissue, fasciae or intramuscular septa. They belonged to deep fibromatosis groups and are characterized by local malignancy because of their propensity for recurrence. They represent less than 0.03% of all tumors and about 3.5% of fibrous tumors [1]. They have survived sporadically or as part of familial adenomatous polyposis (FAP) [2]. Desmoid tumours commonly have a female predominance. They can occur at any age with extreme ages of 15 to 60 years in the literature [3]. Traumatic history, wound scars, implants of the breast, female hormones, and the genetic factors (association with the PAF) have been reported as factors related to the occurrence of desmoid-type fibromatosis [1, 4]. In our case, apart from oral contraception, no other factor has been reported, notably no family history, no trauma or scar. The idiopathic origin has been retained. Clinically, desmoid tumors are usually not painful, often revealed by the palpation of a mass. They can develop in all structures musculo-aponévrotic of the body, but are localized mainly in the abdominal area. We describe 3 forms [5]: desmoid tumors extra-abdominal, essentially interesting the posterior part of the trunk, the chest wall and the lower limbs (58%). Desmoids tumors of the abdominal wall (37%); intra-abdominal tumors invading the pelvis, the blood sugar and tricyclic retinitis (15%). Histologically, these tumors are constituted by a prolifer of fibrous tissue, made of bundles of fibroblasts and abundant collage, without cellular atypia. Their tendency to local invasiveness and recidivism is responsible for their severity. The tumors desmoids of the abdominal wall, although histologically identical to other tumors desmoids, especially extra-abdominal, are particular in their location, their circumstances of occurrence and their therapeutic management [5]. The diagnosis is based on the anatomo-pathological study.
Imaging is contributory to diagnosis. Ultrasound coupled to Doppler is the first examination performed in practice, but not very specific, the scanner is interested in superficial tumors and can guide the biopsy. Magnetic resonance imaging (MRI) remains waiting for choice for deep tumors to better characterize the reports as well as in the monitoring of therapy [6]. In our case the scanner was sufficient to identify the tissue character and to predict the reports before the biopsy. The therapeutic management strategy depends on the resectability of the tumor [6]. The wide surgical excision of the lesion is the treatment of reference. However, the infiltrative character of the tumour may cause difficulties for complete surgical resection which can be associated with an excessive risk of mortality or functional sequelae without the risk of metastasis [5]. Surgical excision in first intention aims at obtaining healthy banks; however, because of the infiltrative nature of the tumor, the rate of local relapse is greater than 90% [7]. For these reasons, surgery may need tobe followed by postoperative radiotherapy to control residual or recurrent disease, despite the benign nature of desmoid tumours [8]. Chemotherapy can be considered for unresectable tumours, orfor patients who are unable to support the morbidity of surgery andradiotherapy [9]. Significant tumour reduction with low-dose methotrexate and vinblastine was reported. Several other agents have also been used in the treatment of desmoid tumours, such as hormonal therapy and non-steroidal anti-inflammatory drugs (NSAID) [10]. Meloxicam, a COX-2 inhibitor NSAID, has been shown to be effective in control ling neck and other extra-abdominal desmoid tumours [10]. Evolution is unpredictable [5]. Spontaneous regression is observed in 10% of cases, stabilization or sarcomatous transformation are also possible. Surveillance should be long-term [5]. The prognosis depends on the age, location and extent of the lesion, but also the number of recurrences. It is better in extra-abdominal locations [5].
Extra-abdominal locations of desmoid tumors are rare. The early diagnosis of these tumors allows for better management and the surveillance must be for long-term because there is a risk of local recurrence.
The authors declare no competing interests.
All the authors contributed in writing this article.
Figure 1: the nodule is poorly limited by 3 cm fixed in the deep plane and mobile at the superficial level without opposing inflammatory signs
Figure 2: abdomino-pelvic computed tomography had revealed a suspicious parietal tumor
Figure 3: A, B) HESx 5; fuso-cellular proliferation in long divergent bundles, generally not very cellular and rich in collagen, often wavy. Medium abundance vascularization; C) HESx10; This proliferation is not very cellular and is available on an abundant collagen background responsible for this undulating aspect of the nuclei; D) HESx40; at high magnification, there are no atypies or mitoses
Figure 4: the tumor expressed anti-smooth muscle actin antibody (AML)
- Montagliani L, Duverger V. Les tumeurs desmoides. J Chir (Paris). 2008;145:20-6. Google Scholar
- Zehani-Kassar A, Ayadi-Kaddour A, Marghli A, Ridene I, Daghfous H, Kilani T et al. Desmoid-type chest wall fibromatosis: a six cases series. Orthopaedics and Traumatology: Surgery and Research. 2011 Feb;97(1):102-7. PubMed | Google Scholar
- Koukoutsis I, Pappas A, Karanikas G. Desmoid tumor of the supra clavicular region: a case report. Cases J. 2009;2:7222. Google Scholar
- Shohei Mori, Yuki Noda, Daiki Kato, Shinichi Hirooka, Takashi Ohtsuka. Desmoid-type fibromatosis arising in a bifid rib chest wall. General Thoracic and Cardiovascular Surgery. 2019 Nov;67(11):996-998. PubMed | Google Scholar
- Aissa A, Alouini-Mekki R, Ben Abdallah A, Enaifar R, Kobbi I, Stita W, Boulifi A. Mise à jour sur la prise en charge des tumeurs desmoides. Gynécologie Obstétrique & Fertilité. 2012 Feb;40(2):104-8. PubMed | Google Scholar
- Ben Haj Amor M, Ploton L, Ceugnart L, Taïeb S. Magnetic resonance imaging of desmoid-type fibromatosis: current evaluation criteria. Bull Cancer. 2020 Mar;107(3):359-363. PubMed | Google Scholar
- Belembaogo E, Kirova YM, Le Bourgeoise JP. Traitement de la tumeur desmoide récidivante inopérable. Med Afr Noire. 2000;47:6. Google Scholar
- Risoud M, Mortuaire G, Leroy X, Leblond P, Fayoux P. Desmoid tumours of the head and neck in children: Review of management. European Annals of Otorhinolaryngology, Head and Neck diseases. 2017 May;134(3):155-160. PubMed | Google Scholar
- Constantinidou A, Jones RL, Scurr M, Al-Muderis O, Judson I. Advanced aggressive fibromatosis: Effective palliation with chemotherapy. Acta Oncol. 2011 Apr;50(3):455-61. PubMed | Google Scholar
- Huang PW, Tzen CY. Prognostic factors in desmoid-type fibromatosis: aclinic pathological and immunohistochemical analysis of 46 cases. Pathology. 2010;42:147-50. Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures

Article metrics
PlumX Metrics
Desmoid tumor of the abdominal wall: case report
