
Primary intestinal lymphangiectasia associated to Epilepsy: a case report
Jallouli Olfa, Ben Nsir Sihem, Bouchaala Wafa, Kamoun Fatma, Triki Charfi Chahnez
Corresponding author: Jallouli Olfa, Department of Pediatric Neurology, Hedi Chaker University Hospital, Sfax Tunisia, LR19ES15 Sfax University, Sfax, Tunisia 
Received: 25 Apr 2021 - Accepted: 18 Jul 2021 - Published: 19 Jul 2021
Domain: Neurology (general)
Keywords: Primary Intestinal Lymphangiectasia, Epilepsy, case report
©Jallouli Olfa et al. PAMJ Clinical Medicine (ISSN: 2707-2797). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Jallouli Olfa et al. Primary intestinal lymphangiectasia associated to Epilepsy: a case report. PAMJ Clinical Medicine. 2021;6:27. [doi: 10.11604/pamj-cm.2021.6.27.29503]
Available online at: https://www.clinical-medicine.panafrican-med-journal.com//content/article/6/27/full
Case report 

Primary intestinal lymphangiectasia associated to Epilepsy: a case report
Primary intestinal lymphangiectasia associated to epilepsy: a case report
![]() Jallouli Olfa1,2,&, Ben Nsir Sihem1,2, Bouchaala Wafa1,2, Kamoun Fatma1,2,
Jallouli Olfa1,2,&, Ben Nsir Sihem1,2, Bouchaala Wafa1,2, Kamoun Fatma1,2, ![]() Triki Charfi Chahnez1,2
Triki Charfi Chahnez1,2
&Corresponding author
Seizures occur frequently in inflammatory diseases. Primary Intestinal Lymphangiectasia (PIL) is a systemic disorder, but epilepsy was not reported before. In this paper, we presented the case of 17 years old boy, diagnosed for PIL, who developed by the age of 16 recurrent epileptic seizures. The epilepsy was confirmed, the patient was treated with carbamazepine with good outcomes. Epilepsy may occur in patients with PIL, and doctors should be aware and treat seizures when the diagnosis of symptomatic acute seizures is excluded.

Primary intestinal Lymphangiectasia (PIL) is a rare disease with unknown etiology. It is responsible for systemic manifestations due to protein-losing enteropathy. Symptomatic acute seizures (SAS) may occur. Hypocalcemia is the major cause of those seizures. However, PIL associated to epilepsy was not described before in published-cases. It is important to differentiate between SAS and epilepsy. Epilepsy is due to unprovoked recurrent seizures; however, SAS is due to an acute disturbance that affects the central nervous system. Anti-epileptic drugs are used in epilepsy to control seizures. In SAS, they remain useless and only the correction of the acute disturbance may control seizures. We present, in this article, a child who developed the symptoms of PIL since the age of one year. Lately, he developed epileptic seizures. Symptomatic acute seizures (SAS) was infirmed and the diagnosis of epilepsy was confirmed. The patient had good outcomes under epileptic drugs.

Patient information: here we report the case of a 17-year-old, followed since the age of 1 year for PIL, and who consulted in emergency for the first paroxysmal phenomena. The child had symptoms of PIL since the age of one year. He had lymphedema of the lower left limb, hydrocele and multiple renal and hepatic cysts. The biological investigations revealed also low levels of protid and albumin in the blood. The diagnosis of PIL was confirmed at the age of 11 years old based on typical histological findings. The biopsy of the duodenum mucosa showed the presence of lymphangiectasia (only reports were available). Since then, he received an albumin transfusion every month and was adherent to a particular diet with satisfying outcomes with no need for other treatments.
Clinical findings: at the age of 16 years, he was admitted in our Pediatric Neurology Department of Sfax University Hospital for one paroxysmal phenomenon during sleep, described as hypertonia of 4 limbs then clonic movements lasting 5 minutes. He dropped from the bed and bitted his tongue. He awaked 15 minutes later and he was confused. He was admitted the same day at the emergency room. His neurological exam was normal, and he had a full-weight development.
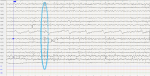
Diagnostic approach: at the admission in the Child Neurology Department, biological tests (Table 1) and cerebral computed tomography (CT) scan were done within 24 hours and were normal. A symptomatic cause for this epileptic seizure was excluded. Electroencephalogram was done within the 24 hours showed repetitive bifrontal spikes diffusing sometimes in posterior region (Figure 1).
Therapeutic intervention and follow-up: the diagnosis of epilepsy was taken, and the patient was treated with Carbamazepine, 10 mg/kg/day, without recurrence of seizures during 7 months; then, he stopped his antiepileptic drug by himself and 5 months later, he had a prolonged generalized clonic seizure which lasted 10 minutes that stopped by intravenous Clonazepam. Biological tests specially calcemia (Table 1) and brain magnetic resonance imaging (MRI) was done and reminded normal. Recurrence of seizures was not due to biological disturbance or cerebral lesion. With the normality of complementary tests, we could exclude the possibility of SAS and strengthen the diagnosis of epilepsy. Carbamazepine was prescribed again with positive outcome during 2 years.
Here, we reported a 17 old boy with PIL syndrome associated to focal epilepsy, with exclusion of symptomatic acute seizures by normal biological tests and brain MRI. In this case, the diagnosis of epilepsy was admitted for many reasons: the recurrence of spontaneous and unprovoked seizures, the epileptiform anomalies in EEG, the well response on antiepileptic drugs and the recurrence of seizures after treatment arrest. PIL can be associated with symptomatic acute seizures due to biological disturbances caused by the loss of protein end electrolytes. So far, a case of an adult with symptomatic acute seizures was published [1]. Antiepileptic drugs were not prescribed and seizures did not repeat after the normalization of biological tests. Our patients had always normal blood tests. The symptomatic acute seizures are due to electrolytes and calcium loss. The PIL is a disease affecting lymphatic system, leading to anasarca, chronic diarrhoea, hypoproteinaemia, lymphoedema and chylous effusions explaining the loss of calcium and electrolytes [2,3]. Sporadic disease was found to account in 90% of cases, like our patient. Familial basis was noted in the remaining 10% of the cases [4]. Intestinal lymphangiectasis is found to be associated with various genetic and acquired diseases [3-5]. Currently, postulated mechanism of lymphedema and protein loss involves pressure abnormalities in lymphatic channels due to congenital anomaly of development or obstruction due to secondary causes. An increase in intraluminal pressure in lymphatics leads to dilatation in the intestinal wall which leads to leakage of protein and consequent hypoproteinemia [5].
The protein-losing enteropathy accounts for anasarca, lymphopenia and immunoglobulin deficiency. Lymphedema and chylous effusions also result from chronic lymphatic leakage [2]. Patients with PIL usually present in the first decade of life with symptoms resulting from chronic protein loss and lymphatic leakage. Edema, diarrhea, malabsorption and poor weight gain are features most commonly documented in previous reports [3,4]. Our patient presented all of these symptoms since the age of one year and the disease was confirmed at the age of 11 after histological result of the duodenum biopsy showing the presence of lymphangiectasia. He was adherent to the treatment and diet and could have normal cognitive, motor and weigh development. Our patient, at the age of 16 consulted for epileptic seizures beginning. All complementary exams were normal including biological investigations and cerebral MRI. The possibility of acute symptomatic seizures was excluded. We could not find an etiology for his epilepsy. It is not described before the association of PIL and epilepsy. Many case reports were published of the disease including other manifestations or the beginning of the disease an elderly patient but none of them described the occurrence of epilepsy in children diagnosed for PIL. The patient has all criteria for epilepsy. In fact, he had non provoked epileptic seizures with positive response to antiepileptic treatment.
We could not find an explication in litterateur for the appearance of epilepsy or a link with PIL, for this young man, so we tried to compare with the other systemic disorders. PIL is a rare disease and publications are almost case reports, that is why, it is not well known all systemic manifestations and their mechanisms. Comparing to the other systemic disorders, the occurrence of seizures may be understood by one of two mechanisms. The first cause results from a diffuse disruption of homeostasis in the body disrupting the brain´s ability to balance excitatory and inhibitory potentials. The second is a result of a focal insult to the brain itself, whether it be through breakdown of the blood-brain barrier, inflammation, or otherwise [6]. The only explication that we propose for our patient is it is probably the systemic inflammation was responsible for the seizures or may be the genetic etiology for the PIL can also be responsible for epilepsy. The patient is observed and satisfied with the treatment.
Patient consent:the patient provided his informed consent.
The combination of malabsorption, anasarca and chylous effusions is a strong pointer towards intestinal lymphangiectasia. Diagnosis requires high degree of suspicion and endoscopic biopsies are sufficient for confirmation. Exclusion of secondary causes of intestinal lymphangiectasia is paramount to the diagnosis of PIL, especially in areas with a high prevalence of tuberculosis and filariasis. Form this case report, we wander if it remains necessary to search epileptic manifestations or epileptic discharges by demanding an electroencephalogram for all patients diagnosed having the disease.
The authors declare no competing interests.
All authors participated in the care of the patient and the writing of the manuscript. All authors read and approved the final version of the manuscript.
Table 1: biological tests done within the first and second admissions in neurology department
Figure 1: EEG recording showing repetitive frontal spikes diffusing on posterior regions
- Harrison's Principles of Internal Medicine, 15th ed. Neurology. 2001;57(10):1941-1941.
- Waldmann T, Steinfeld J, Dutcher T, Davidson J, Gordon R. The role of the gastrointestinal system in “idiopathic hypoproteinemia”. Gastroenterology. 1961;41:197-207. PubMed
- Troskot R, Jurčić D, Bilić A, Gomerčić Palčić M, Težak S, Brajković I. How to treat an extensive form of primary intestinal lymphangiectasia. World J Gastroenterol. 2015;21(23):7320-5. PubMed | Google Scholar
- Vignes S, Bellanger J. Primary intestinal lymphangiectasia (Waldmann´s disease). Orphanet J Rare Dis. 2008;3(1):5. Google Scholar
- McGrath EE, Blades Z, Anderson PB. Chylothorax: aetiology, diagnosis and therapeutic options. Respir Med. 2010;104(1):1-8. PubMed | Google Scholar
- Hoerth MT, Sirven JI. Seizures due to systemic disease. In: neurological disorders due to systemic disease, Oxford, UK: Wiley-Blackwell. 2013;107-26. Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures
Article metrics

