Langerhans cell histiocytosis in children: a case report and brief review of the literature
Zakaria El Ouali, Nisrine Khoubila, Siham Cherkaoui, Mohamed Rachid, Mouna Lamchahab, Meryem Qachouh, Abdellah Madani, Asmaa Quessar
Corresponding author: Zakaria El Ouali, Department of Hematology and Pediatric Oncology-University Hospital of Ibn Rochd, Casablanca, Morocco 
Received: 25 Oct 2019 - Accepted: 05 Nov 2019 - Published: 11 Nov 2019
Domain: Rheumatology,Pediatric oncology
Keywords: Langerhans cell histiocytosis, children, multiple bone lesions
©Zakaria El Ouali et al. PAMJ Clinical Medicine (ISSN: 2707-2797). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Zakaria El Ouali et al. Langerhans cell histiocytosis in children: a case report and brief review of the literature. PAMJ Clinical Medicine. 2019;1:10. [doi: 10.11604/pamj-cm.2019.1.10.20810]
Available online at: https://www.clinical-medicine.panafrican-med-journal.com//content/article/1/10/full
Case report 

Langerhans cell histiocytosis in children: a case report and brief review of the literature
Langerhans cell histiocytosis in children: a case report and brief review of the literature
Zakaria El Ouali1,&, Nisrine Khoubila1, Siham Cherkaoui1, Mohamed Rachid1, Mouna Lamchahab1, Meryem Qachouh1, Abdellah Madani1, Asmaa Quessar1
1Department of Hematology and Pediatric Oncology, University Hospital of Ibn Rochd, Casablanca, Morocco
&Corresponding author
Zakaria El Ouali, Department of Hematology and Pediatric Oncology, University Hospital of Ibn Rochd, Casablanca, Morocco
Langerhans cell histiocytosis (LCH), formerly known as histiocytosis X, is a non-malignant disease involving clonal proliferation of Langerhans cells. It is an orphan disease affecting mainly the child and the young adult. Its etiology is still unknown, and its clinical spectrum is quite wide. Systemic treatment incorporating steroids and cytostatic drugs for at least one year has improved prognosis of multisystem LCH and represents the current standard of care. We report the case of a multifocal bone LCH in a 2-year-old girl affecting the frontal and parietal bones, associated with emphysematous bulla. The diagnosis was confirmed with histological examination. The evolution was favorable after treatment. Based on this observation, we will make a brief review of the literature to present the clinical, histological, radiological, therapeutic and evolutionary aspects of this orphan disease.

Langerhans cell histiocytosis (LCH) is a rare group of disorders whose etiologies are incompletely characterized and understood. Formerly known as histiocytosis X, the disease has a wide spectrum of clinical presentations, ranging from isolated disease with spontaneous resolution to life-threatening multisystem disease [1]. We report the case of a 2-year-old girl who presented a LCH initially affecting the frontal and parietal bones, and secondarily caused emphysematous bulla.

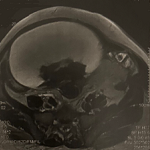
A 2-year-old girl was admitted in the department of hematology and pediatric oncology with a symptomatology of rounded frontal swelling, measuring 3 cm in diameter that was progressively increasing for 3 months (Figure 1). She had a good overall physical condition, particularly normal height and weight regarding her age, no bone pain, no changes in activity level, and no fever. Radiological explorations consisted of a medical ultrasound of the formation, revealing a nodule of the frontal soft tissues, with an osteolytic lesion related to the frontal bone. The cerebral computed tomography (CT) scan showed the presence of a well-defined oval tissue formation in the frontal bone, enhanced after injection of contrast agent, measuring 16.6 x 12.6 mm lysing the opposite bone (Figure 2A). It is associated with a second localization in the left parietal bone, with the same characteristics as the frontal one, measuring 16.1 x 11.3 mm (Figure 2 B). The thoracic CT scan revealed the presence of two subpleural emphysematous bulla located in the right Fowler. A biopsy of the frontal formation was performed, showing bone and fibrous tissues containing a polymorphic cellular infiltrate made of mononucleate histiocytic elements. Some nuclei were kidney-shaped, others were convoluted and grooved (Figure 2 C). This infiltrate was also made of neutrophils and especially of eosinophilic polynuclear cells and lymphocytes. The immunohistochemical study revealed that histiocytic cells diffusely express the CD1a, and focally express the CD68 and PS100. In the same operative time as the biopsy, a myelogram was performed thinking of a possible metastatic neuroblastoma that showed an inflammatory marrow. Work-up of extension consisted of a bone scintigraphy, which showed the absence of a clear scintigraphic sign in favor of distant bone localization. We followed the LCH treatment guidelines (LCH-III protocol) established by the Histiocyte Society (HS). It consisted of an initial 6-week course of therapy with vinblastine (6 mg/m2) and prednisone (40 mg/m2). An assessment at the end of this initial course was performed with a CT scan than showed the persistence of both lytic bone lesions with a net regression of the tissue component. A second course of treatment with vinblastine and prednisone was then undetaken. The assessment made with another CT scan concluded in an almost total regression of the frontal and parietal bone lesions (Figure 3), while the emphysematous bulla were persistent. Maintenance therapy consisted of pulses of vinblastine and prednisone every 3 weeks and daily continuous 6-mercaptopurine for a total treatment duration of 12 months.
Histiocytoses are rare disorders characterized by the accumulation of cells derived from dendritic cells or macrophages. The first classification of histiocytosis, published in 1987 by the working group of the HS, consisted of 3 categories: langerhans cell (LC) or non-LC-related, and malignant histiocytoses (MH). A new classification has been elaborated based on histology, phenotype, molecular alterations, and clinical and imaging characteristics, dividing the histiocytoses into 5 groups resumed in Table 1 [2]. Among histiocytic disorders, LCH is the most common one, affecting an estimated 4 to 5 per million children 0 to 15 years of age each year. The median age of diagnosis is 3.5 years, and the highest incidence rate is observed before 1 year of age, with a decreased incidence observed thereafter [1]. Although major advances have increased our understanding of LCH, its pathophysiology remains unknown and may be the result of either an aberrant autoimmune response or a neoplastic process [1,3]. The most commonly affected organs are: bone (80%), skin (33%), pituitary (25%), liver (15%), spleen (15%), hematopoietic system (15%), lungs (15%), lymph nodes (5-10%), and the central nervous system excluding the pituitary (2-4%) [3]. Systemic signs, such as fever, lethargy, and weight loss, may be noted in patients with either single-system or multisystem disease. Bone involvement is mostly unifocal and most commonly presents as a soft tissue mass presenting with swelling or pain [1], as it was the case in our patient, whose presenting symptom was a soft tissue mass with no pain however. Any bone may be involved, excluding the hands and feet. Radiographically, LCH typically mimics multiple myeloma, presenting as either single or multiple osteolytic lesions; however, in contrast to multiple myeloma, LCH lesions may be accompanied by a periosteal reaction [1]. Skin is the second most frequently involved organ system after bone, lesions may be either circumscribed or spread 77 and there may be either single or multiple lesions [1]. Risk organs include the hematologic system, the spleen and the liver [3]. The lung had been considered for many decades as a risk organ, but in the absence of involvement of other risk organs, lung disease is only in exceptional cases the ultimate cause of death. This usually occurs through ‘‘mechanical complications’’ such as an uncontrolled pneumothorax, or as a late event due to chronic emphysematous changes [3]. The emphysematous bulla were present in our patient from the first CT scan and stayed the same with no clinical expression throughout the evolution of the disease. Many treatments for multisystem LCH, from minimal therapy to intensive chemotherapy, have been tried. However, a randomized trial of treatment made of Vinblastine and etoposide, associated with one dose of corticosteroids, were equally effective treatments for multisystem LCH, but patients who did not respond within 6 weeks were at increased risk for treatment failure and would require different therapy [4]. These evidences have been used to establish the HS evaluation and treatment guidelines that currently represent the standard of treatment. Our patient had a treatment consisting of an initial 6-week course of therapy with vinblastine and prednisone, and the response after the first 6 weeks was a net regression of the tissue component, confirming the results of the study. Regarding the prognosis, for patients older than 2 years at the time of diagnosis, a 10-year survival rate of 97% has been reported; for those younger than 2 years, the rate is 77% [5]. Patients with multisystem disease have an overall survival rate of approximately 71% at 10 years. When at-risk organ systems (spleen, liver, bone marrow, lungs) are involved, 10-year survival decreases to approximately 50% [5].
LCH is a rare entity with various and non-specific clinical presentations. It usually affects bones, but other organs can be involved. The diagnosis of LCH is based on histological and immunophenotypic examination of lesional tissue. Uni or multifocal bone involvement is often of good prognosis. The treatment is controversial, involving chemotherapy and corticotherapy.
The authors declare no competing interests.
Zakaria El Ouali: conceptualization, data curation, investigation, writing, original draft; Nisrine Khoubila: data curation, supervision, validation, writing, review & editing; Siham Cherkaoui, Mohamed Rachid, Mouna Lamchahab, Meryem Qachouh, Abdellah Madani, Asmaa Quessar; supervision. All the authors have read and agreed to the final manuscript.
Table 1: histiocytoses classification showing the 5 groups of diseases
Figure 1: pictures of the frontal swelling in front (A) and side (B) views
Figure 2: initial CT scan images showing the frontal (A) and parietal (B) formations, with the cranial reconstruction (C)
Figure 3: photography of the child’s face after treatment
- Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: diagnosis, differential diagnosis, treatment, sequelae, and standardized follow-up. Journal of the American Academy of Dermatology. 2018;78(6):1035-44. PubMed | Google Scholar
- Emile J-F, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127(22):2672-81. PubMed | Google Scholar
- Haupt R, Minkov M, Astigarraga I, Schäfer E, Nanduri V, Jubran R et al. Langerhans cell histiocytosis (LCH): Guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years: Guidelines for Langerhans Cell Histiocytosis. Pediatric Blood & Cancer. 2013;60(2):175-84. PubMed | Google Scholar
- Gadner H, Grois N, Arico M, Broadbent V, Ceci A, Jakobson A et al. A randomized trial of treatment for multisystem Langerhans’ cell histiocytosis. The Journal of Pediatrics. 2001;138(5):728-34. PubMed | Google Scholar
- Arkader A, Glotzbecker M, Hosalkar HS, Dormans JP. Primary Musculoskeletal Langerhans Cell Histiocytosis in Children: An Analysis for a 3-Decade Period. Journal of Pediatric Orthopaedics. 2009;29(2):201-7. PubMed | Google Scholar
Search

This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures



Article metrics
Recently from the PAMJ-CM