Intestinal ischemia in paroxysmal nocturnal hemoglobinuria with associated aplastic anemia: a rare and challenging association (case report)
Ismail Belefqih, Houda Bachir, Imane Kamaoui, Siham Hamaz, Habiba Alaoui, Khalid Serraj
Corresponding author: Ismail Belefqih, Department of Internal Medicine, Faculty of Medicine and Pharmacy of Oujda, Oujda, Morocco 
Received: 29 Sep 2022 - Accepted: 12 Nov 2022 - Published: 17 Nov 2022
Domain: Gastroenterology,Internal medicine
Keywords: Aplastic anemia, paroxysmal nocturnal hemoglobinuria, intestinal ischemia, complement inhibitors, case report
©Ismail Belefqih et al. PAMJ Clinical Medicine (ISSN: 2707-2797). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Ismail Belefqih et al. Intestinal ischemia in paroxysmal nocturnal hemoglobinuria with associated aplastic anemia: a rare and challenging association (case report). PAMJ Clinical Medicine. 2022;10:31. [doi: 10.11604/pamj-cm.2022.10.31.37585]
Available online at: https://www.clinical-medicine.panafrican-med-journal.com//content/article/10/31/full
Case report 

Intestinal ischemia in paroxysmal nocturnal hemoglobinuria with associated aplastic anemia: a rare and challenging association (case report)
Intestinal ischemia in paroxysmal nocturnal hemoglobinuria with associated aplastic anemia: a rare and challenging association (case report)
![]() Ismail Belefqih1,&,
Ismail Belefqih1,&, ![]() Houda Bachir1, Imane Kamaoui2, Siham Hamaz1, Habiba Alaoui1,
Houda Bachir1, Imane Kamaoui2, Siham Hamaz1, Habiba Alaoui1, ![]() Khalid Serraj1
Khalid Serraj1
&Corresponding author
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare hematological disorder due to an acquired genetic mutation in phosphatidylinositol glycan anchor biosynthesis, class A (PIGA) gene. Paroxysmal nocturnal hemoglobinuria has different manifestations, the most common being hemolysis, thrombosis and aplastic anemia. For the latter, aplastic anemia can be both the cause and the consequence of PNH. Complement inhibitors, such as eculizumab and ravulizumab represents the cornerstone in the management of PNH as it is associated with fewer recurrence of hemolytic and thrombotic events. However, PNH associated aplastic anemia does not improve with complement inhibitor, and is managed either with allogeneic stem cell transplantation or immunosuppressive therapy. The major cause of death in PNH is thrombosis, which is often venous, and of unusual locations. Intestinal ischemia represents a rare location of thrombosis in PNH. There are a few cases reported, where the management was either surgical or conservative. Many of these reports showed a recurrence of thrombosis, and the outcome has been fatal in some. Severe aplastic anemia adds a level of difficulty to the therapeutic challenge as both resection surgery and efficient anticoagulation become infeasible or delicate. We report the case of a 24-year-old woman, who was diagnosed with severe aplastic anemia associated with PNH, for which she was put on triple immunosuppressive therapy. Eight months following the diagnosis, the patient complained of acute abdominal pain with vomiting. A contrast enhanced computed tomography scan showed a jejunal ischemia with signs of perforation. Due to the low platelet level, a conservative approach was taken consisting of oral feeding cessation, hydration, anticoagulation, and antibiotics therapy. This raises the question of whether a complement inhibitor therapy could have prevented this potentially fatal complication.

Paroxysmal nocturnal hemoglobinuria (PNH) is a rare acquired hematological disorder that has 3 classical presentations: hemolysis, thrombosis, and aplastic anemia. While the first two are managed with complement inhibitors, aplastic anemia is managed with triple immunosuppressive therapy or allogeneic stem cell transplant. Thrombosis may represent the major cause of death in PNH [1], but it is rarely reported in association with aplastic anemia, which would make management more challenging.

Patient information: we report the case of a 24-years-old woman, with no underlying comorbidities, who was diagnosed with severe aplastic anemia associated with paroxysmal nocturnal hemoglobinuria (PNH). The diagnosis was made on the basis of a severe pancytopenia, a bone marrow biopsy revealing a very low cellularity, and the detection of a clonal PNH-affected white blood cells at 25 percent. Laboratory results did not show signs of hemolysis, nor did the clinical finding suggest a potential thrombosis. The patient did not have a compatible donor for a potential allogeneic stem cells transplant, and therefore was put on triple immunosuppressive therapy (IST) consisting of cyclosporine at a dose of 150 mg twice a day, eltrombopag 100 mg once a day, and horse antithymocyte globulin which she received four months after the diagnosis, and which was complicated by a hepatic cytolysis that required the discontinuation of eltrombopag for a period of a month until the transaminases levels were back to normal. The cyclosporine serum level was monitored every month and was within the therapeutic range of 200 to 400 ng/mL. Despite the triple IST, there were no signs of improvement as the number and frequency of blood products transfusions remained the same. Eight months following the diagnosis the patient reported an acute diffuse abdominal pain with vomiting.
Clinical findings: physical evaluation found a hemodynamically, respiratory and neurologically stable patient, a mild tachycardia at 110 beats per minute, a fever at 38.2°C, and a tender abdominal palpation with no other clinical findings.
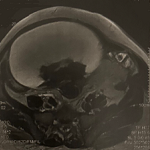
Diagnostic assessment: laboratory findings revealed a pancytopenia with hemoglobin level at 6 g/dl, thrombocytopenia at 14000 elements/mm3, neutropenia at 750 element/mm3, C-Reactive Protein (CRP) level was 250 mg/l, and lipase level was normal. A contrast enhanced computed tomography (CT) scan revealed jejunal ischemia with signs of pre-perforation (Figure 1).
Therapeutic interventions: the patient was managed conservatively with oral feeding stoppage, hydration with isotonic saline, parenteral feeding, anticoagulation with enoxaparin at a dose of 2500 UI once a day and antibiotics consisting of imipenem and metronidazole.
Informed consent: the patient provided a clear and well informed written consent for the publication of the case.
Paroxysmal nocturnal hemoglobinuria is a rare but potentially fatal hematological disorder. It is due to an acquired genetic mutation in phosphatidylinositol glycan anchor biosynthesis, class A (PIGA) gene [2]. The PIGA gene is responsible for the synthesis of glycosylphosphatidylinositol (GPI) anchor, which links proteins to the surface of plasma membranes. CD55 and CD59 are surface cell proteins that inhibit complements and prevent it from attacking the cell. As both the complement inhibitors require a GPI anchor, the mutation in PIGA gene is responsible for a lack of complement inhibitors on cell surface, that manifest as intravascular and extravascular hemolysis [2]. Paroxysmal nocturnal hemoglobinuria can have different manifestations, classical hemolysis, thrombosis, aplastic anemia. For the first two presentations, hemolysis and thrombosis, complement inhibitors, such as eculizumab and ravulizumab have proven their efficacy, unlike in the aplastic anemia presentation where the treatment relies on triple immunosuppressive therapy or allogeneic stem cell transplantation [3].
Only a handful of cases have reported intestinal ischemia in PNH [4-7], most of which were reported in the pre-complement inhibitors era.In these cases, there were two different approaches, while some underwent surgery with resection of necrotic intestinal segments, some used a more conservative approach consisting of oral feeding cessation, hydration, anticoagulation and antibiotics. Both approaches have reported a high risk of recurrence [6,7]. Cases where surgery was performed showed on histological findings hemorrhagic infarcts with focal necrosis [4,5].
Most of these cases were of patients with either hemolytic or thrombotic manifestations that might have required a complement inhibitor therapy. In our case, the patient presentation was purely of a severe aplastic anemia for which she received a triple immunosuppressive therapy. As the major cause of mortality in PNH is thrombosis, this raises the question as whether a complement inhibitors treatment in a severe aplastic anemia might prove beneficial in preventing thrombosis, especially considering the management challenge that aplastic anemia presents as both surgery and anticoagulation become infeasible due to the deep thrombocytopenia.
Paroxysmal nocturnal hemoglobinuria associated aplastic anemia is a disorder where complement inhibitors have not shown efficacy, and where the management relies on either allogeneic stem cell transplant or triple immunosuppressive therapy. However, our case showed that this presentation might still be followed by a thrombotic manifestation, that may be fatal considering its location and the potentially severe thrombocytopenia that may render surgery or anticoagulation infeasible or dangerous. As complement inhibitors have proven their efficacy in preventing thrombosis and hemolysis, their use in aplastic anemia might be beneficial in preventing life threatening ischemia.
The authors declare no competing interests.
Patient management: Ismail Belefqih, Houda Bachir, Imane Kamaoui, Khalid Serraj. Manuscript revision: Houda Bachir, Siham Hamaz, Habiba Alaoui, Khalid Serraj. All the authors have read and agreed to the final manuscript.
Figure 1: abdominal CT scan showing a markedly thickened jejunal wall with discontinued mucosal enhancement
- Brodsky RA. Clinical manifestations and diagnosis of paroxysmal nocturnal hemoglobinuria. UpToDate. 2022. Accessed September 28, 2022.
- Brodsky RA. Pathogenesis of paroxysmal nocturnal hemoglobinuria. UpToDate. 2021. Accessed September 28, 2022.
- Brodsky RA. Treatment and prognosis of paroxysmal nocturnal hemoglobinuria. UpToDate. 2021. Accessed September 28, 2022.
- Ramus J, McPherson GAD. Recurrent bowel infarction in paroxysmal nocturnal haemoglobinuria. J R Soc Med. 2003 Aug;96(8):406-7. PubMed | Google Scholar
- Zapata R, Mella JG, Rollàn A. Intestinal ischemia complicating paroxysmal nocturnal hemoglobinuria. Gastrointest Endosc. 1998 Feb;47(2):184-6. PubMed | Google Scholar
- Doukas MA, Dilorenzo PE, Mohler DN. Intestinal infarction caused by paroxysmal nocturnal hemoglobinuria. Am J Hematol. 1984 Jan;16(1):75-81. PubMed | Google Scholar
- Torres J, De Vroey B, Noël MP, Notteghem B, Colombel JF. Recurrent small bowel ischemia in a patient with paroxysmal nocturnal hemoglobinuria. Nat Rev Gastroenterol Hepatol. 2010 Jul;7(7):410-4. PubMed | Google Scholar
Search

This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures

Article metrics
Recently from the PAMJ-CM