La maladie de Wilson chez l'adulte: à propos de 4 cas
Kaouthar Rais, Ebeibe Dedane, Kaoutar Chahi, Hajar Koulali, Abdelkrim Zazour, Wafaa Khannoussi, Ghizlane Kharrasse, Zahi Ismaili
Corresponding author: Kaouthar Rais, Service d'Hépato-Gastroentérologie, Université Mohammed Premier, Laboratoire de Recherche des Maladies Digestives, Centre Hospitalier Universitaire Mohammed VI, Oujda, Maroc 
Received: 23 Jul 2022 - Accepted: 09 Jun 2023 - Published: 14 Jun 2023
Domain: Hepatology
Keywords: Wilson, survie, cirrhose, cas clinique
©Kaouthar Rais et al. PAMJ Clinical Medicine (ISSN: 2707-2797). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Kaouthar Rais et al. La maladie de Wilson chez l'adulte: à propos de 4 cas. PAMJ Clinical Medicine. 2023;12:16. [doi: 10.11604/pamj-cm.2023.12.16.36354]
Available online at: https://www.clinical-medicine.panafrican-med-journal.com//content/article/12/16/full
Case report 
La maladie de Wilson chez l'adulte: à propos de 4 cas
La maladie de Wilson chez l'adulte: à propos de 4 cas
Wilson's disease in adults: about four cases
![]() Kaouthar Rais1,&, Ebeibe Dedane1, Kaoutar Chahi1, Hajar Koulali1, Abdelkrim Zazour1, Wafaa Khannoussi1, Ghizlane Kharrasse1,
Kaouthar Rais1,&, Ebeibe Dedane1, Kaoutar Chahi1, Hajar Koulali1, Abdelkrim Zazour1, Wafaa Khannoussi1, Ghizlane Kharrasse1, ![]() Zahi Ismaili1
Zahi Ismaili1
&Auteur correspondant
La maladie de Wilson est rare, touchant un enfant pour 30000 naissances. Elle se développe le plus souvent dans la première ou deuxième décennie de la vie. Or, dans cette étude nous rapportons le cas de 4 adultes atteints de la maladie de Wilson avec un âge moyen de diagnostic à 17 ans et un sexe ratio de 1. Le dépistage familial a révélé l´existence de cas similaires chez 9 patients dont seulement 4 sont bien suivis et inclus dans l´étude. Deux patients avaient des signes d´hépatopathie, un patient avait une forme neurologique et un patient était asymptomatique. Le traitement était basé sur la D-pénicillamine, l´évolution sous traitement était favorable chez deux patients avec un décès chez les deux autres et une survie moyenne de 96 mois. On conclut de cette étude que la maladie de Wilson est une maladie rare et grave dont le pronostic reste meilleur chez les patients diagnostiqués à un stade précoce d´où l´intérêt du dépistage familial.
Wilson's disease is rare, affecting one child in every 30,000 births, and most often develops in the first or second decade of life. In this study, we report the case of 4 adults with Wilson's disease, with a mean age of 17 years at diagnosis and a sex ratio of 1. Family screening revealed similar cases in 9 patients, but only 4 of them were well monitored and included in the study. Two patients had signs of liver disease, one patient had a neurological disorder and one patient was asymptomatic. Treatment was based on D-penicillamine: two patients had a favorable outcome, two patients died, with a mean survival of 96 months. Wilson's disease is a rare and serious disease. Early diagnosis is associated with a better prognosis, hence the importance of family screening.
Key words: Wilson, survival, cirrhosis, case report

La maladie de Wilson est une maladie génétique rare à transmission autosomique récessive due à la mutation du gène ATP7B localisé sur le chromosome 13 qui code pour une protéine transporteuse du cuivre. Elle se caractérise par l´accumulation du cuivre essentiellement dans le foie, le système nerveux central et la cornée. Elle touche généralement les patients âgés entre 5 et 35 ans. Les principales manifestations cliniques sont les signes liés à l´atteinte hépatique et les signes neuropsychiatriques. Il existe de rares cas de maladie de Wilson exprimés à un âge adulte. Nous rapportons 4 cas de maladie de Wilson diagnostiqués chez des adultes, durant la période allant de 2015 à 2022 au service d´hépato-gastroentérologie d´Oujda.

Observation n�1
Informations du patient: patient âgé de 18 ans, fils unique, est admis pour la prise en charge de troubles neurologiques notamment une dysarthrie et des troubles de la marche associée à des œdèmes des membres inférieurs.
Résultats cliniques: il avait une ascite avec un syndrome parkinsonien.
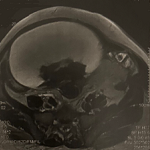
Démarche diagnostique: le bilan biologique a révélé une anémie hémolytique avec test de Coombs négatif, une thrombopénie à 117000 UI/l, TP bas à 45%, hypoalbuminémie à 24g/l et facteur V effondré à 35%. Sur l´échographie abdominale, il avait un foie d´hépatopathie chronique avec des signes d´hypertension portale et une ascite de faible abondance. L´examen ophtalmologique n´a pas objectivé la présence de l´anneau de Kayser-Fleischer et sur l´IRM encéphalique, on notait la présence d´une anomalie de signal de la substance blanche superficielle et profonde en bilatéral, associés à une atteinte des noyaux gris centraux (Figure 1). Le bilan de cuivre a montré une hypercuprurie de 24h à 67g/l et une diminution de la céruloplasmine sérique <0,03g/l. Le score de Leipzig était à 6.
Interventions thérapeutiques: le patient a été mis sous D-pénicillamine 1200mg/jour et Pyridoxine 50mg/jour.
Suivi et résultat: nous avons noté une bonne amélioration sur le plan clinique et biologique. La durée de survie est calculée à 36 mois.
Consentement éclairé: le patient nous a donné son consentement éclairé pour la publication de son cas.
Observation n�2
Informations du patient: patiente âgée de 18 ans, issue d´une fratrie de 3 dont un âgé de 14 ans est porteur de la maladie de Wilson, de découverte fortuite par le dépistage familial. La patiente a consulté pour des hématémèses.
Résultats cliniques: patiente stable sur le plan hémodynamique et respiratoire, les examens abdominal, neurologique et ophtalmologique étaient sans anomalie.
Démarche diagnostique: sur le plan biologique, elle avait une pancytopénie arégénérative avec un TP bas à 50%, une hypercuprurie et une diminution de la céruloplasmine à 0,13g/l faisant un score de Leipzig à 4 en faveur de la maladie de Wilson. Sur la fibroscopie oeso-gastro-duodénale, elle avait des signes endoscopiques d´hypertension portale avec des varices œsophagiennes stade I et II avec signes rouges et une gastropathie hypertensive. L´échographie abdominale a révélé un foie d´hépatopathie chronique avec un tronc porte dilaté et une splénomégalie.
Interventions thérapeutiques: la patiente a été mise sous D-pénicillamine 1200mg/jour.
Suivi et résultat: amélioration clinico-biologique et une durée de survie sans complications calculée à 80 mois.
Consentement éclairé: la patiente est consentante pour la publication de son cas clinique après avoir été avisé de l'intérêt scientifique de partager sa pathologie rare.
Observation n�3
Informations du patient: patient âgé de 17 ans. Le dépistage familial a révélé la présence de quatre malades dans une fratrie de sept dont trois sont décédés à l´âge de 15 ans, 9 ans, 20 jours respectivement et une fille de 4 ans non encore dépistée.
Résultats cliniques: le patient était asymptomatique sur le plan clinique. L´examen ophtalmologique et neurologique étaient sans anomalie.
Démarche diagnostique: sur le bilan biologique, la fonction hépatique était préservée avec une perturbation du bilan de cuivre en faveur de la maladie de Wilson avec un score de Leipzig à 4. L´échographie abdominale était normale.
Interventions thérapeutiques: le patient a été mis sous D-pénicillamine 1200mg/jour avec une mauvaise observance thérapeutique.
Suivi et résultat: l´évolution a été marquée par l´installation d´une insuffisance hépatocellulaire aigüe secondaire à l´arrêt du traitement avec un syndrome de défaillance multi-viscérale responsable du décès du patient, la durée de survie était calculée à 60 mois.
Consentement éclairé: les parents du patient nous ont donné leur consentement éclairé.
Observation n�4
Informations du patient: patient âgé de 16 ans, troisième fils dans une fratrie de six dont deux sont décédés âgés respectivement de 18 ans et 17 ans, une sœur de 18 ans mal suivie, deux frères âgés de 13 ans et 11 ans non dépistés et une sœur de 24 ans saine. Le patient a consulté pour la prise en charge d´une distension abdominale.
Résultats cliniques: il était pâle avec une ascite de grande abondance et une circulation veineuse collatérale.
Chronologie: le Tableau 1 résume la chronologie des événements des 4 patients.
Démarche diagnostique: sur le plan biologique, il avait une anémie hémolytique à 3g/dl, TP bas à 25% et hypoalbuminémie à 15g/l. Sur l´échographie abdominale, le foie était hétérogène dysmorphique de contours irréguliers avec une ascite de grande abondance. Le bilan étiologique a révélé une augmentation du cuivre urinaire de 24h avec diminution de la céruloplasmine; score de Leipzig était à 5, confirmant la maladie de Wilson.
Interventions thérapeutiques: il a été mis sous D-pénicillamine et il a été candidat à une transplantation hépatique.
Suivi et résultat: quatre mois après le diagnostic de sa pathologie, le patient est décédé par un choc hémorragique.
Consentement éclairé: la mère du patient nous a donné son consentement éclairé.
La maladie de Wilson est une affection rare qui touche toutes les races et les ethnies. Sa prévalence moyenne est une personne sur 30 000 à 40 000 dans le monde entier [1]. Certaines populations ont une incidence plus élevée de la maladie de Wilson en raison du taux accru de mariages consanguins. L´âge habituel de diagnostic est de 5 à 45 ans, mais il existe de rares cas détectés chez des enfants de 3 ans et des adultes de 70 ans [2]. Dans notre série, la moyenne d´âge de nos patients était de 17 ans avec un sexe ratio à 1. La maladie de Wilson est une maladie autosomique récessive due à une mutation de l´ATP7B altérant à la fois l´incorporation du cuivre dans la céruloplasmine et l´excrétion du cuivre dans la bile, ce qui est responsable de l´accumulation du cuivre essentiellement dans le foie, le système nerveux central et la cornée. Les principales manifestations cliniques de la maladie de Wilson sont: 1) les signes neurologiques: des tremblements, des troubles de la marche, des mouvements choréiformes, le syndrome parkinsonien, une dysarthrie, une paralysie pseudobulbaire, une dystonie rigide, des convulsions, crise d´épilepsie, migraines et insomnie; 2) les signes ophtalmiques comprennent l´anneau de Kayser-Fleischer (KF) et cataractes en fleur de tournesol; 3) les signes psychiatriques: la dépression, les névroses, les changements de personnalité, les psychoses et l´échec scolaire...; 4) les manifestations hépatiques: ictère, hépatosplénomégalie, ascite, hémorragie digestive, insuffisance hépatocellulaire, hépatite aigüe.
Selon la littérature, les symptômes hépatiques se manifestent entre 10 et 13 ans. Environ 3% seulement de patients au-delà de la quatrième décennie, présentent une maladie hépatique ou neurologique. Les patients diagnostiqués jusqu´à la huitième décennie avaient des symptômes variables allant de tests hépatiques légèrement anormaux à une insuffisance hépatique sub-fulminante [3]. En effet, ils ont noté également que les hommes avaient majoritairement une atteinte neuropsychiatrique (75% contre 58%) et moins d´atteinte hépatique (25% contre 41%). Dans notre série, un patient âgé de 18 ans a consulté pour une hémorragie digestive, l´autre âgé de 16 ans avait une ascite, un seul patient de 18 ans avait des signes neuropsychiatriques et le dernier était asymptomatique. En général, la combinaison d´anneaux de Kayser-Fleischer et d´un taux de céruloplasmine faible (<0,1 g/L) est suffisante pour établir le diagnostic [4]. En l´absence de ses deux arguments, un score de diagnostic « Leipzig » basé sur des critères cliniques, paracliniques et génétiques, a été proposé lors de la 8e réunion internationale sur la maladie de Wilson en 2001 [5]. Ce score était supérieur à 4 chez tous nos patients, ce qui confirme le diagnostic de la maladie de Wilson.
Le traitement de la maladie de Wilson a comme objectif d´éliminer l´excès de cuivre dans l´organisme en favorisant l´excrétion du cuivre par des agents chélateurs tels que la D-pénicillamine et la trientine, et en bloquant l´absorption intestinale du cuivre par des sels de zinc. D´autre part, les aliments à forte teneur en cuivre (le chocolat, les fruits secs, le foie, les graines de sésame, l´huile de sésame, ainsi que les champignons et les noix) doivent être évités [6]. La transplantation hépatique est souvent nécessaire pour les patients se présentant par un tableau d´insuffisance hépatocellulaire inaugurale ou secondaire à l´arrêt du traitement ainsi que les patients cirrhotiques décompensés qui ne répondent pas au traitement médical [7]. En revanche, l´indication de la transplantation hépatique chez les patients ayant des signes neurologiques sévères reste encore débattue [8]. Selon une revue systémique récente englobant 48 articles sur la transplantation hépatique chez les patients présentant des symptômes neurologiques, on note une amélioration majeure de la symptomatologie dans 215 cas (71,2%), une aggravation neurologique dans 24 cas (7,9%), 29 décès (9,6%), et 13 patients (4,3%) ont été perdus de vue. Ces résultats suggèrent que la transplantation hépatique est une méthode prometteuse de prise en charge de la maladie de Wilson chez les patients présentant des symptômes neurologiques sévères, en particulier si le patient n´a pas répondu à un traitement pharmacologique [9].
Dans notre série, on note une nette amélioration clinico-biologique sous traitement. Cependant deux patients sont décédés, l´un d´une insuffisance hépatocellulaire secondaire à l´arrêt du traitement médical et l´autre d´un choc hémorragique. La survie médiane de nos patients sous traitement est calculée à 96 mois avec une survie meilleure sans complication estimée à 142 mois chez le patient diagnostiqué à un stade précoce (Tableau 1). Il n´existe aucun consensus international sur le rythme de surveillance des patients sous chélateur. Selon les recommandations de l´association américaine des maladies hépatiques, ils préconisent une surveillance régulière par un examen physique, le dosage de la céruloplasmine sérique, de la protéinurie et curpurie de 24h, le bilan hépatique ainsi que la numération formule sanguine au moins deux fois par an [10]. L´analyse de l´excrétion urinaire de cuivre sur 24 heures sous traitement médical est utile pour contrôler l´observance du traitement. Les patients prenant la D-pénicillamine ou la trientine doivent avoir une excrétion urinaire de cuivre sur 24 heures de 200 à 500 g/jour (3 à 8 mol/jour); pour les patients sous zinc, elle ne devrait pas dépasser 75 g/jour (1,2 mol/jour).
Dernièrement, un score (King's score) pour déterminer le pronostic des patients a été développé, puis modifié par Dhawan et al. prenant en considération le taux de la bilirubine, ASAT, INR, les globules blancs et l´albumine. Un score supérieur à 11 est en faveur d´un pronostic mauvais de la maladie sans transplantation hépatique [4]. Vu que la maladie de Wilson se transmet sur un mode autosomique récessif, il est primordial de dépister les familles et de rechercher la consanguinité parentale. Dans la présente étude, les quatre familles avaient des frères et des sœurs affectés. Soixante pourcent (60%) des frères et sœurs dépistés avaient également la maladie de Wilson. Nos résultats suggèrent l´importance du dépistage familial pour les patients atteints de la maladie de Wilson.
A travers ces cas cliniques, on déduit d´une part que la principale manifestation clinique des adultes atteints de la maladie de Wilson est l´atteinte hépatique et d´autre part, on présume que le diagnostic de la maladie de Wilson à un âge adulte peut être fatal d´où l´intérêt du dépistage familial qui demeure à nos jours le meilleur moyen diagnostic des formes asymptomatiques se caractérisant par un pronostic meilleur.
Les auteurs ne déclarent aucun conflit d´intérêts.
La prise en charge des patients: Zahi Ismaili, Ghizlane Kharrasse, Wafaa Khannoussi, Abdelkrim Zazour et Hajar Koulali. Collecte des données: Kaouthar Rais, Ebeibe Dedane et Kaoutar Chahi. Rédaction du manuscrit: Kaouthar Rais. Révision du manuscrit: Zahi Ismaili. Tous les auteurs ont lu et approuvé la version finale du manuscrit.
Tableau 1: durée de suivi des patients atteints de la maladie de Wilson
Figure 1: image par résonnance magnétique (IRM) cérébrale en coupe axiale séquence T1 montrant une anomalie de signal de la substance blanche en bilatéral
- Mouzari Y, Abdelkhalek R, El Asri F, Reda K, Oubaaz A. La maladie de Wilson: � propos d'un cas familial [Wilson's disease: about a family case]. Pan Afr Med J. 2014 Aug 2;18:270. PubMed | Google Scholar
- Hermann W. Classification and differential diagnosis of Wilson's disease. Ann Transl Med. 2019 Apr;7(Suppl 2):S63. PubMed | Google Scholar
- Guindi M. Wilson disease. Semin Diagn Pathol. 2019 Nov;36(6):415-422. PubMed
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Wilson's disease. EASL. 2012; 56(3):671-85. Google Scholar
- Ferenci P, Caca K, Loudianos G, Mieli-Vergani G, Tanner S, Sternlieb I, Schilsky M, Cox D, Berr F. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003 Jun;23(3):139-42. PubMed | Google Scholar
- Fernando M, Mourik I Van, Wassmer E, Kelly D. Wilson disease in children and adolescents. BMJ. 2020; 105(5):499-505. PubMed | Google Scholar
- Palumbo CS, Schilsky ML. Clinical practice guidelines in Wilson disease. Ann Transl Med. 2019;7(S2):S65-S65. PubMed | Google Scholar
- Catana AM, Medici V. Liver transplantation for Wilson disease. World J Hepatol. 2012;4(1):5-10. PubMed | Google Scholar
- Litwin T, Bembenek J, Antos A, Przybyłkowski A, Skowrońska M, Kurkowska-Jastrzębska I, et al. Liver transplantation as a treatment for Wilson's disease with neurological presentation: a systematic literature review. Acta Neurol Belg. 2022;122(2):505-518. PubMed | Google Scholar
- Roberts EA, Schilsky ML. Diagnosis and Treatment of Wilson Disease: An Update. AASLD Pract Guidel. 2008;47(6):2089-111. PubMed | Google Scholar
Search

This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures

Article metrics
PlumX Metrics
La maladie de Wilson chez l'adulte: à propos de 4 casRecently from the PAMJ-CM