Atrophie multi-systématisée: à propos d’un cas et revue de la littérature
Francois Kouda, Saydou Ly, Meriem Haloua, Badreeddine Alami, Mustapha Maaroufi, Meryem Boubbou, Youssef Lamrani
Corresponding author: Francois Kouda, Service de Radiologie, CHU Hassan II Fès, Fes, Maroc 
Received: 22 Feb 2020 - Accepted: 28 Feb 2020 - Published: 10 Mar 2020
Domain: Radiology
Keywords: Atrophie multi-systématisée, imagerie
©Francois Kouda et al. PAMJ Clinical Medicine (ISSN: 2707-2797). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Francois Kouda et al. Atrophie multi-systématisée: à propos d’un cas et revue de la littérature. PAMJ Clinical Medicine. 2020;2:90. [doi: 10.11604/pamj-cm.2020.2.90.21912]
Available online at: https://www.clinical-medicine.panafrican-med-journal.com//content/article/2/90/full
Case report 
Atrophie multi-systématisée: à propos d’un cas et revue de la littérature
Atrophie multi-systématisée: à propos d´un cas et revue de la littérature
Multiple system atrophy: about a case and literature review
Francois Kouda1,2,&, Saydou Ly1,2, Meriem Haloua1,2, Badreeddine Alami, Mustapha Maaroufi1,2, Meryem Boubbou1,2, Youssef Lamrani1,2
1Service de Radiologie, CHU Hassan II Fès, Fès, Maroc, 2Faculté de Médecine, Université Sidi Mohammed Benabdellah, Fès, Maroc
&Auteur correspondant
Francois Kouda, Service de Radiologie, CHU Hassan II Fès, Fès, Maroc
L'atrophie multisystématisée (AMS) est une affection neurodégénérative de l'adulte, d'évolution progressive et de pronostic sévère, affectant les voies pyramidales, extrapyramidales, cérébelleuses ainsi que le système nerveux autonome. Les critères diagnostiques sont essentiellement cliniques permettant le diagnostic d'AMS avec deux degrés de certitude: « possible » et « probable »; le caractère certain étant apporté par l'examen anatomopathologique. L´imagerie en particulier, l'IRM cérébrale peut montrer une atrophie putaminale, pontique et des pédoncules cérébelleux moyens. Nous rapportons le cas d´une femme, âgée de 56 ans, atteinte d´AMS dont l´évolution fut marquée par l´installation progressive d´un syndrome démentiel sévère. L´examen neuro-pathologique confirma le diagnostic d´AMS et révéla la présence de marqueurs histologiques de maladie d´Alzheimer. Cette association nosologique est extrêmement rare dans la littérature. Cette observation confirme que la survenue d´une démence sévère au cours de l´évolution d´une AMS doit faire rechercher une cause associée et souligne l´utilité d´une vérification anatomique en cas d´atypie clinique. L´intérêt de notre travail réside dans la rareté de la pathologie.
Multiple system atrophy (MSA) is a neurodegenerative disorder occurring in adults. It is characterized by progressive evolution and severe prognosis. It affects the pyramidal, extrapyramidal and cerebellar system, as well as the autonomic nervous system. Diagnostic criteria are essentially based on clinical examination which allows two degrees of certainty for the diagnosis of MSA: “possible" and "probable"; the degree of diagnostic certainty is established by anatomo-pathological examination. Diagnostic imaging, especially brain MRI, can show putaminal atrophy as well as atrophy of the pontem and of the middle cerebellar peduncles. We report the case of a 56-year old woman with MSA whose evolution was marked by progressive cognitive impairment that led to severe dementia. Neuropathological examination confirmed the diagnosis of MSA and revealed the presence of histological Alzheimer´s markers. This nosologic association has been extremely rarely reported in the literature. This study confirms that the occurrence of severe dementia in patients with MSA requires assessment of an associated cause and highlights the importance of anatomical examination in the case of clinical abnormality. The interest of our study lies in the rarity of the disease.
Key words: Multiple system atrophy, diagnostic imaging

L'atrophie multisystématisée (AMS) est une pathologie neurodégénérative rare, sporadique, survenant le plus souvent dans la sixième décennie avec une possible prédominance masculine même si cette dernière est discutée [1]. Elle appartient avec la maladie de Parkinson et la démence à corps de Lewy, à la famille des synucléinopathies [2]. Les critères diagnostiques sont essentiellement cliniques permettant le diagnostic d'AMS avec deux degrés de certitude : « possible » et « probable » ; le caractère certain étant apporté par l'examen anatomopathologique [1]. Le tableau clinique est typiquement dominé par une atteinte cérébelleuse, un syndrome parkinsonien akinétorigide, une dysfonction pyramidale et une dysautonomie [2]. Certains examens complémentaires permettent d'apporter des arguments pour le diagnostic d'AMS comme la présence d'anomalies évocatrices à l'IRM cérébrale ou la présence d'une dénervation dopaminergique au Dat-Scan en cas d'AMS [1]. Son pronostic est plus sombre avec une survie médiane inférieure à 10 ans [3].

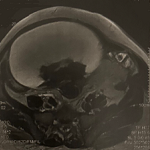
Patiente de 56 ans amenée en consultation pour dysarthrie et trouble de la marche, vertige, des acouphènes et des nausées d´aggravation progressive. Devant le syndrome cérébelleux et vestibulaire le neurologue a demandé une IRM du crane prenant les CAI, à la recherche de l´étiologie. L´IRM a permis de mettre en évidence des anomalies de signal du mésencéphale, et des noyaux dentelé associées à une atrophie cérébelleuse et pontique (Figure 1). L´association des signes cliniques et IRM a permis de poser le diagnostic de l´atrophie multi systématisée.
La première description clinique et histologique d´atrophie olivo-ponto-cérébelleuse (AOPC) sporadique est attribuée à Dejerine et Thomas en 1900. En 1960, Adams et al ont décrit des patients présentant un syndrome parkinsonien lié à une dégénérescence combinée de la substance noire et du striatum, syndrome qu´ils nommèrent dégénérescence striato-nigrique (DSN). Mais, c´est seulement en 1969 que Graham et Oppenheimer ont proposé le terme d´atrophie multisystematisée (AMS) pour regrouper ces trois différents syndromes cliniques émanant d´un même processus dégénératif [4]. L´atrophie multisystématisée (AMS) est une maladie dégénérative sporadique rare. Elle est cependant le syndrome parkinsonien atypique le plus fréquent avec la paralysie supranucléaire progressive (PSP). Elle est classée parmi les “synucléinopathies”, comme la maladie de Parkinson idiopathique (MPI) et la démence à corps de Lewy (DCL) [2,5]. La prévalence de l´AMS varie de 2 à 5 cas pour 100 000 habitants. La maladie débute le plus souvent dans la 6e décennie, quelle que soit la forme. Il existe une légère prédominance masculine (1,3 à 1,9 homme/1 femme) [1,2,4].
La physiopathologie de l'AMS reste mal connue et on ne peut pas encore définir avec certitude les raisons pour lesquelles les cellules se détériorent dans les cas d´AMS. L´origine de la maladie est probablement poly-factorielle. Des recherches en cours se penchent toutefois sur l´exposition à des toxines environnementales ou à des traumatismes importants. Bien que la présence d'antécédents familiaux soit un critère d'exclusion pour le diagnostic d'AMS, plusieurs cas au sein de même familles ont été rapportées relançant l'hypothèse d'une part génétique [1]. Le diagnostic d'AMS repose avant tout sur des critères cliniques consensuels [1,6] au sein desquels des anomalies spécifiques IRM ont été identifiées [1]. Etablis en 1998, ils ont été récemment revus lors d´une nouvelle conférence de consensus. Selon ces critères consensuels, il existe deux types d´AMS : l´AMS-P dans laquelle le syndrome parkinsonien prédomine et l´AMS-C où le syndrome cérébelleux est au premier plan. Par ailleurs, on classe la maladie selon trois niveaux de certitude: AMS «possible», «probable » et «certaine». L´AMS «possible» se définit par un syndrome parkinsonien ou un syndrome cérébelleux, et un signe suggérant une dysautonomie associée à un autre signe clinique évocateur ou une anomalie de neuro-imagerie. L´AMS « probable » se définit par une maladie neurologique progressive, débutant à l´âge adulte, incluant rigoureusement une dysautonomie définie (c´est-à-dire une incontinence urinaire associée à une dysfonction érectile chez l´homme, ou une hypotension orthostatique survenant dans les 3 minutes du lever avec chute de la pression artérielle d´au moins 30 mmHg pour la systolique ou de 15 mmHg pour la diastolique) et un syndrome parkinsonien avec une réponse pauvre à la Lévodopa ou un syndrome cérébelleux. La certitude diagnostique est obtenue à l´examen anatomopathologique post mortem qui met en évidence une dégénérescence des structures olivo-ponto-cérébelleuses et de la voie nigrostriée, associée à d´abondantes inclusions gliales intra cytoplasmiques d´α-synucléine [4,7].
L´IRM cérébrale et l´imagerie fonctionnelle peuvent apporter des arguments supplémentaires pour le diagnostic d´AMS [5]. On peut ainsi observer, dans l´AMS, une atrophie putaminale, pontique et des pédoncules cérébelleux moyens. Sur les séquences pondérées en T2, on peut souvent noter un hyposignal de la partie postérieure du putamen, parfois associée à un hyper signal de la bordure postéro-latérale du putamen, ainsi qu´un hyper signal en forme de «croix» pontique et plus rarement des hyper signaux floconneux des pédoncules cérébelleux moyens. La séquence T2* ou T2 Echo Gradient semble détecter de façon plus précoce l´hyposignal putaminal lié à un dépôt de fer. Cet hyposignal putaminal est nettement plus fréquent dans l´AMS que dans les autres syndromes parkinsoniens, même s´il n´est pas spécifique. En pratique, la séquence T2* peut permettre d´aider au diagnostic devant un syndrome parkinsonien répondant mal à la Lévodopa. En outre, l´IRM de diffusion semble permettre de dissocier de façon plus spécifique les AMS des maladies parsoniennes idiopathiques. L´IRM cérébrale peut être cependant normale au début de maladie. La tomographie par émission de positons (TEP) avec le (18F)-fluorodésoxyglucose peut mettre en évidence un hypo métabolisme du cervelet ou du striatum. Chez un patient présentant un syndrome parkinsonien atypique sans syndrome cérébelleux, la démonstration par TEP d´un hypo-métabolisme cérébelleux permet le diagnostic d´une AMS-P « possible ». De même, chez un patient présentant un syndrome cérébelleux sans syndrome parkinsonien, l´observation d´une dénervation dopaminergique nigrostriée par tomographie d´émission monophotonique (TEMP) ou TEP est en faveur d´un diagnostic d´AMS-C « possible ». Toutefois, les examens par TEP ne sont pas actuellement des examens de routine [1,4,5]. Les traitements médicamenteux du syndrome parkinsonien, mais surtout du syndrome cérébelleux, restent décevants. En revanche, le traitement symptomatique de la dysautonomie, cardiovasculaire et vésico-sphinctérienne, peut améliorer la qualité de vie des patients. L´accompagnement, le soutien psychologique, la prise en charge sociale et ergothérapique sont tout aussi importants pour améliorer les conditions de vie des patients AMS [4] C´est une maladie de mauvais pronostic. La moitié des patients est en fauteuil roulant après 5 ans, et la médiane de survie moyenne est de 8 à 10 ans selon les études [5].
L´AMS est une affection neurodégénérative rare de mauvais pronostic. Le diagnostic reste essentiellement clinique. Cependant, le développement de l´IRM cérébrale conventionnelle avec des séquences spécifiques et de l´imagerie fonctionnelle pourra, dans l´avenir, aider le clinicien à rechercher plus précocement les signes radiologiques en faveur d´une AMS possible.
Les auteurs ne déclarent aucun conflit d´intérêts.
J´ai rédigé ce manuscrit avec le soutient mes collègues Amina Alaoui, Saydou Ly. Meriem Haloua, Badreeddine Alami, Youssef Lamrani, Mustapha Maaroufi, Meryem Boubbou, mes professeurs ont corrigé le manuscrit avant soumission. Tous les auteurs ont lu et approuvé la version finale du manuscrit.
Figure 1: IRM cérébrale coupes axiales Flair, diffusion, 3D T1 après injection de Gadolinium, coronale T2: Hypersignal T2, Flair des 02 pédoncules cérébelleux moyens, du mésencéphale et hyposignal des noyaux dentelés non rehaussé après contraste et non restrictif en diffusion. Il s'y associe une atrophie cérébelleuse et pontique
- Foubert-Samier A, Tison F, Meissner WG. L'atrophie multisystématisée. Pratique Neurologique - FMC. 2015;6(2):115-123. Google Scholar
- Rusina R, Bourdain F, Matej R. Atrophie multi-systématisée et maladie d'Alzheimer: association rare de deux affections neurodégénératives; À propos d´un cas. RevNeurol (Paris). 2007;163 (12):1239-1241. Google Scholar
- Wenning GK, Geser F, Krismer F, Seppi K, Duerr S, Boesch S et al. The natural history of multiple system atrophy: a prospective European cohort study. Lancet Neurol. 2013;12(3):264-74. PubMed | Google Scholar
- Nathalie Damon-Perrière, François Tison, Wassilios Meissner. L´atrophie multisyste'matise'e. Psychol NeuroPsychiatr Vieil. 2010; 8(3):179-91. Google Scholar
- Nathalie Damon-Perrière, François Tison, Wassilios Meissner. Atrophie multi systématisée: nouveaux critères diagnostiques. La Lettre du Neurologue. 2009; 13:10. Google Scholar
- Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71(9):670-6. PubMed | Google Scholar
- Trojanowski JQ, Revesz T. Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. NeuropatholApplNeurobiol. 2007 Dec;33(6):615-20. PubMed | Google Scholar
Search

This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words

Article metrics
Recently from the PAMJ-CM