Manifestations oto-vestibulaires inaugurales révélant un syndrome de Gougerot-Sjögren primitif: observation clinique et revue diagnostique
Ahmed Rouihi, Oumaïma Mansoum, Dina Rayane Benariba, Mohamed Zalagh, Saloua Ouraini, Bouchaib Hemmaoui, Fouad Benariba, Noureddine Errami
Corresponding author: Ahmed Rouihi, Service d'Oto-rhino-laryngologie et de Chirurgie Cervico-faciale, Hôpital Militaire d'Instruction Mohamed V, Rabat, Maroc 
Received: 26 Oct 2025 - Accepted: 26 Nov 2025 - Published: 09 Feb 2026
Domain: Otolaryngology (ENT)
Keywords: Syndrome de Gougerot-Sjögren, vertige, oreille interne, auto-immunité, surdité neurosensorielle, atteinte vestibulaire, corticothérapie
Funding: Ce travail n'a reçu aucune subvention spécifique d'un organisme de financement des secteurs public, commercial ou à but non lucratif.
©Ahmed Rouihi et al. PAMJ Clinical Medicine (ISSN: 2707-2797). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Ahmed Rouihi et al. Manifestations oto-vestibulaires inaugurales révélant un syndrome de Gougerot-Sjögren primitif: observation clinique et revue diagnostique. PAMJ Clinical Medicine. 2026;20:2. [doi: 10.11604/pamj-cm.2026.20.2.49970]
Available online at: https://www.clinical-medicine.panafrican-med-journal.com//content/article/20/2/full
Case report 

Manifestations oto-vestibulaires inaugurales révélant un syndrome de Gougerot-Sjögren primitif: observation clinique et revue diagnostique
Manifestations oto-vestibulaires inaugurales révélant un syndrome de Gougerot-Sjögren primitif: observation clinique et revue diagnostique
Initial otovestibular manifestations revealing primary Gougerot-Sjögren syndrome: a clinical case and diagnostic review
Ahmed Rouihi1,&, Oumaïma Mansoum1, Dina Rayane Benariba1, Mohamed Zalagh1, Saloua Ouraini1, Bouchaib Hemmaoui1, Fouad Benariba1, Noureddine Errami1
&Auteur correspondant
Le syndrome de Gougerot-Sjögren (SGS) est une maladie auto-immune systémique touchant principalement les glandes exocrines. Les atteintes oto-vestibulaires, bien que rares, peuvent précéder les manifestations glandulaires typiques, rendant le diagnostic initial difficile. Nous rapportons le cas d'une patiente de 71 ans présentant des vertiges progressifs, des acouphènes bilatéraux et des céphalées diffuses. Les explorations audiovestibulaires ont objectivé une surdité neurosensorielle bilatérale légère associée à une hypofonction vestibulaire modérée. L'imagerie par résonance magnétique (IRM) cérébrale révélait des hypersignaux punctiformes de la substance blanche, compatibles avec une atteinte neurologique du syndrome de Gougerot-Sjögren. Le bilan immunologique a mis en évidence la positivité des anticorps anti-SSA/Ro et la biopsie des glandes salivaires accessoires a confirmé le diagnostic de syndrome de Gougerot-Sjögren primitif. Ce cas illustre une présentation inaugurale rare du SGS révélée par des troubles vestibulaires isolés. Il souligne l'importance d'évoquer une étiologie auto-immune devant tout vertige inexpliqué, afin de permettre un diagnostic et une prise en charge précoces.
Gougerot-Sjögren syndrome (GSS) is a systemic autoimmune disease that primarily affects the exocrine glands. Although rare, otovestibular involvement may precede typical glandular manifestations, making initial diagnosis challenging. We report the case of a 71-year-old woman presenting with progressive vertigo, bilateral tinnitus, and diffuse headaches. Audiovestibular examinations revealed mild bilateral sensorineural hearing loss associated with moderate vestibular hypofunction. Magnetic resonance imaging (MRI) of the brain revealed white matter hyperintensities (WMH), consistent with neurological involvement in Gougerot-Sjögren syndrome. Immunological testing revealed positive anti-SSA/Ro antibodies, and a minor salivary gland biopsy confirmed the diagnosis of primary Gougerot-Sjögren syndrome. This case illustrates a rare initial presentation of GSS revealed by isolated vestibular symptoms. It highlights the importance of considering an autoimmune etiology in all cases of unexplained vertigo in order to enable early diagnosis and appropriate management.
Key words: Gougerot-Sjögren syndrome, vertigo, inner ear, autoimmunity, sensorineural hearing loss, vestibular involvement, corticosteroid therapy

Le syndrome de Gougerot-Sjögren (SGS) est une maladie auto-immune systémique caractérisée par une infiltration lymphoplasmocytaire des glandes exocrines, responsable de xérostomie et de xérophtalmie [1]. Au-delà des manifestations glandulaires classiques, des atteintes extra-glandulaires, notamment neurologiques et oto-vestibulaires, peuvent précéder les signes sicca, retardant le diagnostic [2]. Les troubles auditifs et vestibulaires incluent surdité neurosensorielle, acouphènes et vertiges, et peuvent constituer les premières manifestations d'une atteinte auto-immune de l'oreille interne [3,4]. Nous rapportons une observation clinique illustrant un vertige inaugural révélant un SGS primitif, et discutons les aspects diagnostiques et thérapeutiques à la lumière des données actuelles.

Information du patient: une patiente de 71 ans, sans antécédents pathologiques particuliers, a consulté pour des vertiges d'installation progressive évoluant depuis plusieurs semaines. Ces épisodes étaient accompagnés d'acouphènes bilatéraux et de céphalées diffuses, sans autres symptômes neurologiques associés.
Antécédents médicaux et familiaux: aucun antécédent d'otite, de traumatisme crânien, de maladie auto-immune ou d'exposition à des agents ototoxiques n'a été rapporté. Il n'existait pas d'antécédent familial de surdité ni de pathologie auto-immune.
Examen clinique: l'examen otologique retrouvait des conduits auditifs externes normaux et des tympans intacts. L'examen neurologique était globalement normal, hormis un discret déséquilibre à la marche. Aucun nystagmus spontané, paralysie faciale ou autre anomalie neurologique n'a été observé.
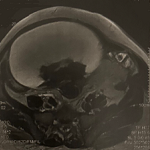
Investigations cliniques et paracliniques: l'audiométrie tonale liminaire objectivait une surdité neurosensorielle bilatérale légère, prédominant sur les hautes fréquences, avec une discrimination vocale conservée (Figure 1). Les explorations vestibulaires (VNG et vHIT) mettaient en évidence une hyporéflexie bilatérale modérée, en faveur d'une atteinte vestibulaire périphérique. L'imagerie par résonance magnétique (IRM) cérébrale révélait des hypersignaux punctiformes de la substance blanche, compatibles avec une atteinte neurologique du syndrome de Gougerot-Sjögren (Figure 2). Le bilan biologique standard était sans particularité. Le bilan immunologique montrait une positivité des anticorps anti-SSA/Ro et anti-SSB/La. Le test de Schirmer objectivait une sécheresse oculaire modérée. Enfin, la biopsie des glandes salivaires accessoires mettait en évidence une sialadénite lymphocytaire de grade IV selon Chisholm et Mason, confirmant le diagnostic de syndrome de Gougerot-Sjögren primitif.
Intervention thérapeutique: une corticothérapie orale à base de prednisone (1 mg/kg/jour, dégressive sur six semaines) a été instaurée, associée à un programme de rééducation vestibulaire comprenant des exercices d'équilibre et d'adaptation visuelle.
Suivi et résultats: après quatre semaines de traitement, la patiente rapportait une nette amélioration de la stabilité posturale et une diminution des acouphènes. Aucun épisode de vertige récidivant n'a été noté au suivi à six mois.
Consentement éclairé: le consentement écrit de la patiente a été obtenu pour la publication de cette observation clinique.
Le syndrome de Gougerot-Sjögren (SGS) est une affection auto-immune chronique, caractérisée par une infiltration lymphoplasmocytaire des glandes exocrines, entraînant une xérostomie et une xérophtalmie typiques [1]. Cependant, dans certains cas, des manifestations extra-glandulaires, notamment oto-vestibulaires, peuvent précéder les signes sicca, rendant le diagnostic particulièrement difficile [2,3]. Ces atteintes restent sous-diagnostiquées, car elles sont souvent attribuées à des causes périphériques ou à des vertiges idiopathiques.
Sur le plan physiopathologique, plusieurs mécanismes ont été proposés pour expliquer les atteintes de l'oreille interne au cours du SGS. L'hypothèse la plus largement admise est celle d'une atteinte auto-immune directe médiée par des anticorps anti-SSA/Ro et anti-SSB/La dirigés contre des antigènes de la strie vasculaire et du ganglion spiral, entraînant une altération de la microcirculation cochléovestibulaire [4,5]. L'infiltration lymphocytaire périvasculaire et la vascularite des petites artères labyrinthiques peuvent provoquer une ischémie des structures sensorielles de l'oreille interne, responsable d'une surdité neurosensorielle ou d'un déficit vestibulaire [6]. D'autres études ont suggéré l'existence d'un phénomène inflammatoire diffus comparable à celui observé dans les maladies auto-immunes de l'oreille interne, où la production d'auto-anticorps et de cytokines pro-inflammatoires induit une atteinte bilatérale progressive [7,8].
Cliniquement, les atteintes oto-vestibulaires dans le SGS se manifestent par une triade variable: surdité neurosensorielle, acouphènes et vertiges [5]. Dans plusieurs séries, jusqu'à 25% des patients atteints de SGS présentaient une hypoacousie, souvent asymétrique et prédominant sur les hautes fréquences [6]. L'atteinte vestibulaire, bien que plus rare, peut être inaugurale comme dans notre cas. Elle se traduit par une hyporéflexie bilatérale au VNG ou au vHIT, pouvant simuler une vestibulopathie bilatérale idiopathique. L'IRM cérébrale permet d'éliminer une origine centrale, notamment une sclérose en plaques ou une vascularite du tronc cérébral, tandis que la confirmation biologique par la positivité des anticorps anti-SSA/SSB et la biopsie des glandes salivaires reste déterminante pour établir le diagnostic [2,9]. Cette combinaison d'arguments cliniques, immunologiques et histopathologiques permet de distinguer le SGS d'autres causes de vertiges auto-immuns ou métaboliques.
Le traitement repose essentiellement sur une approche immunomodulatrice précoce. La corticothérapie systémique demeure le traitement de première ligne, avec une amélioration clinique rapportée dans 60 à 70% des cas [8]. En cas de résistance ou de rechute, le recours à des immunosuppresseurs tels que l'azathioprine, le méthotrexate ou le mycophénolate mofétil peut être envisagé [7]. Le rituximab, anticorps monoclonal anti-CD20, a montré une efficacité prometteuse dans certaines formes réfractaires d'atteinte auditive ou vestibulaire liée au SGS [10]. Parallèlement, la rééducation vestibulaire occupe une place essentielle dans la récupération fonctionnelle, en stimulant les mécanismes d'adaptation et de compensation centrale [9].
Le pronostic dépend essentiellement de la précocité du diagnostic et du traitement. Les atteintes vestibulaires isolées, bien que rares, peuvent régresser sous corticothérapie, mais les formes prolongées exposent à un risque de séquelles fonctionnelles irréversibles. Dans notre observation, la réponse favorable à la corticothérapie et à la rééducation souligne l'intérêt d'une reconnaissance rapide de ces manifestations atypiques.
En définitive, cette observation illustre l'importance d'évoquer une cause auto-immune, notamment un SGS primitif, devant tout vertige inexpliqué, en particulier chez les sujets âgés ou de sexe féminin. Une approche multidisciplinaire associant ORL, internistes et immunologistes permet non seulement un diagnostic plus précoce mais aussi une meilleure prévention des séquelles auditives et vestibulaires à long termse.
Le SGS peut se manifester initialement par des troubles vestibulaires ou auditifs précédant les signes sicca. La réalisation d'un bilan audiovestibulaire complet, d'un profil immunologique et d'une biopsie des glandes salivaires est indispensable pour le diagnostic. La corticothérapie systémique et la rééducation vestibulaire constituent les piliers du traitement.
Les auteurs ne déclarent aucun conflit d'intérêts.
Tous les auteurs ont participé à la conduite du travail, à la rédaction du manuscrit et ont approuvé la version finale pour soumission.
Figure 1: audiométrie tonale liminaire montrant une surdité neurosensorielle bilatérale légère et symétrique chez une patiente atteinte de syndrome de Gougerot-Sjögren primitif
Figure 2: imagerie par résonance magnétique cérébrale montrant des hypersignaux punctiformes de la substance blanche, évocateurs d´une atteinte neurologique du syndrome de Gougerot-Sjögren
- Mavragani CP, Moutsopoulos HM. Primary Sjögren's syndrome. N Engl J Med. 2014;370:2410-2420.
- Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE et al. Classification criteria for Sjögren's syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis. 2002;61:554-558. PubMed | Google Scholar
- Bovo R, Ciorba A, Martini A. Autoimmune inner ear disease: review of the literature and new perspectives. Autoimmun Rev. 2011;10:628-632.
- Gidley PW, Coleman HT. Cochlear microangiopathy and autoimmune inner ear disease. Laryngoscope. 2013;123:1245-1250.
- Mandl P et al. Audiovestibular involvement in primary Sjögren's syndrome: a clinical series. Clin Rheumatol. 2010;29:1239-124.
- Chatzistefanou KI et al. Vestibular dysfunction in Sjögren's syndrome: prevalence and clinical features. Rheumatology (Oxford). 2012;51:1183-1188.
- Fox RI. Sjögren's syndrome. Lancet. 2005 Jul 23-29;366(9482):321-31. PubMed
- Arslan S et al. Corticosteroid therapy in autoimmune inner ear disease associated with Sjögren's syndrome. Audiol Neurootol. 2005;10:81-87.
- Herdman SJ, Clendaniel RA. Vestibular Rehabilitation. 4th ed. Philadelphia: F.A. Davis; 2014. Google Scholar
- Bacciu A et al. Rituximab in autoimmune inner ear disease: case reports and literature review. Rhinology. 2011;49:64-68.
Search

This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures


Article metrics
Recently from the PAMJ-CM