Oncocytic adrenal adenoma: a case report
Omar Iraqui Houssaini, Younes Houry, Moussaab Rachid, Ghassane El Omri, Abdeljalil Heddat
Corresponding author: Omar Iraqui Houssaini, Department of Urology, Cheikh Khalifa International University Hospital, Mohammed VI University of Sciences and Health (UM6SS), Casablanca, Morocco 
Received: 19 Jan 2026 - Accepted: 07 Feb 2026 - Published: 19 Feb 2026
Domain: Urology
Keywords: Tumor, adrenal, anatomopathological, oncocytes, case report
Funding: This work received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
©Omar Iraqui Houssaini et al. PAMJ Clinical Medicine (ISSN: 2707-2797). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Omar Iraqui Houssaini et al. Oncocytic adrenal adenoma: a case report. PAMJ Clinical Medicine. 2026;20:10. [doi: 10.11604/pamj-cm.2026.20.10.51180]
Available online at: https://www.clinical-medicine.panafrican-med-journal.com//content/article/20/10/full
Case report 

Oncocytic adrenal adenoma: a case report
Oncocytic adrenal adenoma: a case report
Omar Iraqui Houssaini1,&, Younes Houry1, Moussaab Rachid1, ![]() Ghassane El Omri1, Abdeljalil Heddat1
Ghassane El Omri1, Abdeljalil Heddat1
&Corresponding author
Oncocytic adrenal tumors are a rare entity among adrenal neoplasms. Common in women, they are characterized by a proliferation of large cells with eosinophilic and granular cytoplasm. They are found in various organs such as the salivary glands, kidneys, parathyroid glands, and, very rarely, the adrenal glands. Often presenting with subtle and misleading clinical symptoms, they are frequently characterized by vague abdominal pain or lower back pain with a deterioration in general health. We report the case of a 65-year-old female patient who presented with the sudden onset of left-sided low back pain, which had been paroxysmal in nature and accompanied by headaches, palpitations, and profuse sweating for several years. The interest of this case report is to highlight this rare entity and raise interest in the importance of a multidisciplinary approach combining imaging, endocrine evaluation, and histopathological analysis to assess the diagnosis.

Adrenocortical oncocytic adenoma, or adrenal oncocytoma, is a rare entity among adrenal neoplasms. It currently accounts for less than 1% of these neoplasms [1,2]. It predominantly affects women, and oncocytomas are frequently found in the kidney, salivary glands, and thyroid, but very rarely in the adrenal gland [3]. It is characterized by the presence of large cells with eosinophilic, granular cytoplasm rich in mitochondria, giving them a distinctive histological appearance [4]. We report the case of a 65-year-old female patient with diabetes and hypertension who presented at a urology clinic with the sudden onset of left-sided low back pain, which had been paroxysmal in nature and accompanied by headaches, palpitations, and profuse sweating for several years.

Patient information: we present the case of a 65-year-old female patient with a medical history of untreated high blood pressure, diabetes controlled with oral antidiabetic drugs (OADs), and who had undergone a tubal ligation 10 years ago. She presented to the urology clinic with a sudden onset of diffuse lower back pain, without nausea or vomiting. The patient also reported frequent headaches, palpitations, and profuse sweating. All of these symptoms occurred in the context of a deterioration in her general condition and a weight loss of 8 kilograms.
Clinical findings: the clinical examination revealed a surgical scar above the pubic bone and painful left lumbar contact on palpation; the rest of the clinical examination was unremarkable.
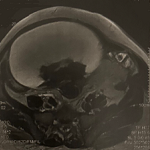
Ultrasound and laboratory assessment: an ultrasound scan was performed during the consultation, revealing an oval-shaped, well-defined, avascular mass measuring 40 x 48mm in the left suprarenal tissue. Uro-CT scan revealed the presence of an oval formation appearing to be dependent on the left adrenal gland, hypodense, heterogeneously enhanced after contrast injection, measuring 57 x 35 mm (Figure 1). The scan also revealed a normal-sized left kidney with regular contours and no focal abnormalities, secreting and excreting within physiological limits. No other lesions or lymphadenopathy were detected in the adjacent organs. As an adrenal origin was strongly suspected, the patient's radiological results were supplemented by biological tests, which revealed preserved renal function with urea at 0.27 and creatinine at 7.11. The hemostasis assessment was unremarkable. Endocrinological assessments revealed an increased aldosterone/renin ratio of 45.1 and a greatly increased standing ratio of 100.6. Measurement of methoxylated catecholamine derivatives showed an elevated level of free metanephrine at 67.7 ng/L, a level of free normetanephrine at 39.9 ng/L, and 3-methoxytyramine at 42.7 ng/L. No difficulties in accessing care were encountered in this specific case.
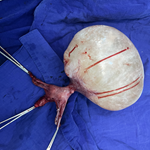
Timeline: in view of the clinical context and the results of radiological and biological examinations, the patient underwent laparoscopic adrenalectomy. This was preceded by close monitoring of blood pressure and treatment with alpha blockers at a gradually increasing dose 14 days before surgery. The adrenalectomy (Figure 2) was technically challenging due to difficult access to the adrenal gland, requiring rigorous control of bleeding and careful dissection of neighboring structures. The procedure was also marked by a brief catecholaminergic crisis during mobilization of the gland, a classic immediate complication requiring rapid hemodynamic stabilization.
Diagnostic assessment: the pathological findings initially revealed a tumour proliferation of epithelial appearance with a diffuse, alveolar, trabeculocordonal architecture and nests of medium-sized polygonal cells. The finely granular eosinophilic cytoplasm contained ovoid nuclei showing marked cytonuclear atypia in places. Endocrine-like stroma. Mitoses were estimated at 9 mitoses per 10 high-power fields, with the presence of atypical mitoses. No foci of tumour necrosis or vascular invasion were observed. The capsule appeared intact, with no capsular invasion, and the interface with the adjacent adrenal parenchyma remained clear, giving the lesion a well-circumscribed appearance (Figure 3). Additional immunohistochemical testing showed positivity for anti-Melan A antibody, anti-Inhibin antibody, Calretinin, and anti-vimentin. Immunolabeling with anti-Melan-A antibody and anti-Inhibin-α antibody showed diffuse cytoplasmic expression within the tumor proliferation, with moderate to intense and relatively homogeneous expression intensity (Figure 4, Figure 5). Immunomarking with anti-calretinin antibody similarly shows focal cytoplasmic and nuclear expression within the tumor proliferation, predominantly at the periphery of certain tumor nodules and involving a limited number of neoplastic cells. The intensity of the marking appears to be low to moderate and heterogeneous.
In the overall morphological and immunohistochemical context already documented, combining the expression of steroidogenic markers such as inhibin-α and Melan-A, this focal positivity reinforces the argument in favor of adrenal differentiation. It also helps to rule out the main non-adrenocortical differential diagnoses, particularly neuroendocrine tumors and metastases of other origins (Figure 6). Furthermore, immunohistochemistry did not detect any anti-synaptophysin antibody positivity. Ki-67 was assessed at 1%, with nuclear positivity limited to rare cells scattered within the tumor proliferation. The staining appears focal and non-diffuse, with no areas of proliferative hyperactivity or identifiable hot spots (Figure 7).
Final diagnosis: in this tumor context, it is consistent with the other morphological elements, in particular the absence of necrosis, capsular invasion, and vascular invasion, and helps to reinforce the classification of the tumor in the oncocytic adenoma group according to the Lin-Weiss-Bisceglia criteria [5]. The immunohistochemical profile concluded that there was an oncocytic adrenal adenoma.
Follow-up and outcomes: the postoperative course was uneventful; postoperative assessments were unremarkable. Postoperative cardiological follow-up was performed, finding no abnormalities on ECG or TEE. No adjuvant treatment was given. The patient was stable in terms of hemodynamics, blood pressure, and respiration, and was hospitalized for seven days. Follow-up was arranged, including a clinical examination and abdominal ultrasound every six months for the first year, then annually for the following five years.
Patient perspective: “I am so grateful to the medical professionals who were able to diagnose the cause of my suffering and my unexplained weight loss. The procedure was fairly quick.”
Informed consent: written informed consent was obtained from the patient and can be provided upon request.
Oncocytic adrenal adenoma is now a rare condition [2], usually discovered during radiological examination and confirmed by pathological examination of the adrenalectomy [1]. First described in the 1950s, fewer than 200 cases of adrenal oncocytoma have been reported in the scientific literature. Predominantly affecting women, oncocytomas are often located in the kidneys, parathyroid glands, and salivary glands, but rarely in the adrenal glands [3,6]. Adrenal oncocytic adenomas sometimes have atypical radiological characteristics, which make it difficult to distinguish them from adrenal cortical carcinoma or an indeterminate lesion. They are generally well-defined, with heterogeneous enhancement after contrast injection, and are often larger than 4 cm. They are described as a collection of large cells in histology, polygonal and granular with abundant eosinophilic cytoplasm, finely granular, linked to a massive accumulation of mitochondria. These tumors, which are often well-defined, can be benign (oncocytic adenomas), of uncertain malignant potential, or malignant (oncocytic carcinomas) [4].
The classification of this entity is now based on the Lin-Weiss-Bisceglia score, which refers mainly to major and minor criteria. Major criteria include a high mitotic index (>5 mitoses/field), the presence of atypical mitoses, and venous, capsular, or sinusoidal invasion. Minor criteria include tumor size greater than 10 cm and/or weight greater than 200 grams, the presence of tumor necrosis, and diffuse cellular atypia [5,7]. The classification of adrenal neoplasms is also based on Ki-67, a marker of cell proliferation rate that is considered a major prognostic parameter in the management of these tumors [8].
The histological study is generally supplemented by immunohistochemistry to support the diagnosis. Immunohistochemically, oncocytic adrenal cortical tumors have a characteristic profile marked by the expression of adrenal cortex markers, notably SF-1, Melan-A, inhibin-α, and calretinin, confirming their steroidogenic differentiation [7]. Strong positivity for anti-mitochondrial antibodies reflects the abundance of organelles specific to oncocytic cells, while the absence of expression of neuroendocrine markers, such as chromogranin A and synaptophysin, allows the main differential diagnoses, such as pheochromocytoma, to be ruled out. The evaluation of the Ki-67 proliferation index is also an essential complementary factor: generally low in benign oncocytic adenomas (5%), it helps to refine the distinction with forms of uncertain malignant potential or oncocytic carcinomas, where higher values may be observed [5].
Our case study was based on a 65-year-old female patient with known diabetes and hypertension who was admitted for urology consultation for the sudden onset of left low back pain accompanied by headaches, palpitations, and profuse sweating. Radiological examinations revealed an oval formation appearing to be dependent on the left adrenal gland, hypodense, heterogeneously enhanced after contrast injection, and measuring 57 x 35 mm. Laboratory tests revealed increased levels of methoxylated catecholamine derivatives and abnormal endocrine function, with an increased aldosterone/renin ratio in both standing and sitting positions. Left laparoscopic adrenalectomy was indicated in accordance with the latest recommendations. Close blood pressure monitoring was implemented in view of the planned surgery, along with alpha-blocker treatment due to suspected catecholamine secretion. Alpha-adrenergic blockade prevents potentially severe blood pressure spikes and hemodynamic instability induced by manipulation of the gland during surgery.
The adrenalectomy proved technically challenging due to the deep and sometimes limited access to the adrenal gland. Removal of the gland was also marked by a transient episode of catecholaminergic hyperdischarge, an immediate complication well described during adrenal manipulation, requiring increased anesthetic vigilance and rapid hemodynamic stabilization. Despite these difficulties, the excision was completed under good conditions.
The pathological findings then revealed a 60-gram specimen measuring 5.2 cm in length, showing diffuse, alveolar, trabecular-corded epithelial tumor proliferation. Histological cross-sections revealed clusters of medium-sized polygonal cells with eosinophilic, finely granular cytoplasm and ovoid nuclei showing cytonuclear atypia. Mitoses were estimated at 9 mitoses per 10 high-power fields, with the presence of a few atypical mitoses. Furthermore, the absence of tumor necrosis, vascular invasion, and capsular invasion was noted. Complementary immunohistochemistry confirmed the adrenal cortical origin of the tumor, with diffuse expression of SF-1, Melan-A, Vimentin, Calretinin, and Inhibin-α. The abundant positivity to anti-mitochondrial antibodies was consistent with oncocytic differentiation. Despite the symptoms described by the patient, which were consistent with Menard's triad, neuroendocrine markers, particularly chromogranin and synaptophysin, were negative, allowing pheochromocytoma or endocrine tumor to be formally ruled out [8]. All of these histological results, together with the immunohistochemical profile and a low Ki-67 proliferation index, were consistent with a diagnosis of benign oncocytic adrenal adenoma.
Oncocytic adrenal adenoma is a rare condition whose clinical and radiological presentation can be misleading, posing a real diagnostic challenge. This case highlights the importance of a multidisciplinary approach combining imaging, endocrine evaluation and histopathological analysis to establish a definitive diagnosis. Adrenalectomy remains the standard treatment, allowing both a definitive diagnosis and curative management, with a generally favorable outcome when the criteria for benignity are confirmed.
The authors declare no competing interests.
Omar Iraqui Houssaini and Younes Houry: both authors contributed equally and are considered co-first authors. All the authors have read and approved the final version of this manuscript.
The authors thank the patient for consenting to this publication.
Figure 1: A, B) CT scan findings demonstrating an oval-shaped mass appearing to arise from the left adrenal gland
Figure 2: the surgical specimen from the adrenalectomy
Figure 3: hematoxylin-eosin-stained histological section showing an oncocytic adrenocortical adenoma at low, medium, and high magnification
Figure 4: immunohistochemical section showing positive expression of inhibin-α
Figure 5: immunohistochemical section showing positive expression of melan-A
Figure 6: immunohistochemical section showing positive staining for calretinin
Figure 7: immunohistochemical section showing a low Ki-67 proliferation index
- Cardona Attard CD, Gauci Z, Gatt N, Scicluna W, Cachia MJ. Oncocytic adrenocortical tumour presenting as an incidentaloma: a diagnostic challenge. BMJ Case Rep. 2022;15(9):e250900. PubMed | Google Scholar
- Kakimoto S, Yushita Y, Sanefuji T, Kondo A, Fujishima N, Kishikawa M et al. Non-hormonal adrenocortical adenoma with oncocytoma-like appearances. Hinyokika Kiyo. 1986;32(5):757-763. PubMed | Google Scholar
- Smirnova EA, Mikhalov IG. Electron microscopic characteristics of oncocytoma of the lung, small intestine and adrenal gland. Arkh Patol. 1986;48(6):79-81. PubMed | Google Scholar
- Cao L, Dong X, Tan J, Wang Z. Adrenocortical oncocytic neoplasms: a case report. Front Med (Lausanne). 2025;12:1709401. PubMed | Google Scholar
- Mete O, Erickson LA, Juhlin CC, de Krijger RR, Sasano H, Volante M, et al. Overview of the 2022 WHO Classification of Adrenal Cortical Tumors. Endocr Pathol. 2022;33(1):155-196. PubMed | Google Scholar
- Kanitra JJ, Hardaway JC, Soleimani T, Koehler TJ, McLeod MK, Kavuturu S. Adrenocortical oncocytic neoplasm: A systematic review. Surgery. 2018;164(6):1351-1359. PubMed | Google Scholar
- Bisceglia M, Ludovico O, Di Mattia A, Ben-Dor D, Sandbank J, Pasquinelli G et al. Adrenocortical oncocytic tumors: report of 10 cases and review of the literature. Int J Surg Pathol. 2004;12(3):231-243. PubMed | Google Scholar
- Sada A, Joseph J, Alsibai R, Habermann EB, Foster TR, Lyden ML et al. Oncocytic Adrenal Neoplasms: Clinical Profiles and Long-Term Outcomes. Am Surg. 2025;91(3):381-385. PubMed | Google Scholar
Search

This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures







Article metrics
PlumX Metrics
Oncocytic adrenal adenoma: a case reportRecently from the PAMJ-CM